




18. The History of Thalidomide
No drug has had a greater effect than thalidomide on the extent and intensity of the preclinical investigation of potential medicines required by the regulatory authorities. Indeed, the establishment of thalidomide as the cause of the apparent epidemic of children born with horrific deformities in the late 1950s was responsible for the institution of some regulatory bodies, such as the United Kingdom’s Committee on the Safety of Drugs, and for the strengthening of others, such as the Food and Drugs Administration (FDA) of the United States. Despite this, the history of the development of thalidomide, and of the subsequent studies of the teratogenic and other effects of the drug, has become confused, because of misrepresentation by those anxious to discredit the contribution of animal experimentation to medical advances.
Two categorical statements can be made about thalidomide. First, thalidomide was never administered to pregnant animals before it was used in humans. Secondly, only five months after the teratogenic effects of thalidomide had been established and the drug withdrawn, embryopathic actions of thalidomide were shown to occur in rat and rabbit (1). Over the following ten years fetal malformations caused by thalidomide were demonstrated in seven other species of small mammal and eight species of monkey (2).
Early Experimental Studies
The first paper describing the pharmacological actions of thalidomide was published in 1956 by Kunz, Keller and Mückter from the Research Laboratories of the German pharmaceutical firm Chemie Grünenthal (3). Thalidomide, designated then as K17, was alleged to reduce spontaneous movement in mice without the initial excitement phase observed with other sedative drugs such as phenobarbitone and glutethimide. The onset of action of thalidomide was rapid and the sedative effect more profound and of longer duration of action than that of the comparator drugs. Coordination, as detected by ability of mice to cling to a slowly rotating rod, was not reduced even with doses in excess of those that produced sedation. Of greater significance, thalidomide was claimed to be virtually non-toxic, oral doses in excess of 5000mg/kg failing to cause death, whereas 600mg/kg and 300mg/kg doses of glutethimide and phenobarbitone respectively were sufficient to kill half of the mice in a test group. Chronic administration of 100-500mg/kg thalidomide to mice, rats, guinea pigs and rabbits for 30 days appeared to be well-tolerated. Thus it was proposed by the researchers at Chemie Grünenthal that thalidomide would be a useful sedative or hypnotic that did not carry the suicide risk of contemporary medicines.
Of significance, the authors emphasised that thalidomide was poorly water soluble but claimed, with no evidence, that the apparent innocuity is “not only due to the fact that the compound is sparingly soluble in water; it also indicates extremely low toxicity.” Crucially, no measurements of blood or tissue concentrations of thalidomide in treated animals were performed. Such data would have demonstrated that the lack of toxicity of thalidomide was indeed due to lack of absorption of the drug. The paper by Kunz, Keller and Mückter that appeared in Arzneimittel-Forschung was immediately followed by a clinical report by Jung (4) describing the sedative effects of thalidomide in 300 patients who were given 25-200 mg thalidomide three times a day. There was no comparison with a placebo group and no indication of the duration of treatment was given. The blood picture of 20 of the patients was alleged to be unchanged over 4 weeks and liver function tests in 20 patients with enlarged livers showed no abnormality. However, no numerical data was provided to support these claims. With high doses side effects included sleepiness, giddiness and constipation. Some thirty years after the publication of these papers, the pediatrician Widukind Lenz, who played a significant part in the exposure of thalidomide as the cause of embryopathy, stated: “The papers published in 1956 by Kunz et al. on animal experiments and by Jung on clinical experiences with thalidomide have so little scientific value that in my opinion they should not have been accepted for print.” (5)
The actions of thalidomide were also investigated by G. B. Somers (6), chief pharmacologist at the Distillers Company, which was licensed by Grünenthal to distribute thalidomide in the British and Commonwealth markets. Somers, using thalidomide as a suspension in 1% carboxymethylcellulose, generally confirmed the inhibition of spontaneous movement by the drug in mice and also remarked on its apparent lack of toxicity in animals. However, Somers added an important caveat when discussing the results of his toxicity studies: “It may well be that the absence of toxicity is due to a limited absorption, for the compound has a low solubility in body fluids, and when administered parenterally remains at the site of injection. In the absence of a suitable assay method absorption studies have not yet been made.”
Somers was later proved correct. Some months after his initial experiments he tested the actions of thalidomide prepared as a finely ground suspension in sugar solution. Such a preparation had just been marketed by Grünenthal as a sedative Contergan Saft that allegedly could be safely used by children. Somers was shocked to discover that microfined thalidomide mixed with sugar solution, unlike the suspensions originally used, was highly toxic to mice (7).
Even by the standards of the time, the preclinical and clinical investigation of thalidomide was superficial. This is illustrated by the animal studies performed upon meprobamate, a muscle relaxant and sedative agent first described by Berger in 1954 (8). Acute toxicity was measured in rats after oral administration and intraperitoneal injection, and in mice via these routes as well as after intravenous injection. Acute toxicity was also measured in monkeys. Subacute toxicity was examined in dogs given 1 g of meprobamate daily for between 60-75 days. The blood picture, urine analysis and kidney function tests were performed before administration of the drug and 30 and 60 days after treatment; blood urea nitrogen and urea clearance were measured at 60 days. At autopsy, dogs showed no abnormalities of kidney, stomach, small intestine, liver, bladder or adrenal glands. Chronic toxicity studies were performed over 15 months on five groups of 20 male and female rats. Two groups were used as controls, the remaining groups received 2%, 1% or 0.5% meprobamate in the diet. At the 12th week, male and female animals were mated and the resulting neonates examined for normality. There were no differences between control and test groups in the intensive examinations of the blood and urine, and the histological examination of all the major organs at autopsy. Unlike the relatively superficial preclinical examination of thalidomide reported two years later, Berger looked at the fate of meprobamate in the body, examining the urine for breakdown products. Approximately 10% of the drug was excreted unchanged in the urine, a much greater percentage excreted in a conjugated form, partly as an unknown metabolite conjugated with glucuronide.
Clinical Usage
On the basis of the limited experimental studies, and further clinical trials (none double blind), thalidomide was launched in Germany in November, 1957 as a novel “non-toxic” sedative under the trade name Contergan, although it had had limited use as a component, together with quinine, phenacetin, salicylate and vitamin C, of an anti-influenza preparation termed Grippex since November 1956 (9).
Contergan was marketed aggresively as a completely harmless sedative, and as a result its sales increased markedly throughout 1959. However, its alleged lack of toxicity soon came into question. In October 1959 a neurologist, Dr R Voss diagnosed polyneuritis in three patients who had taken Contergan for a year, and raised with Grünenthal his concern that the drug might have a toxic action on peripheral nerves. Despite the fact that, from April 1959, Grünenthal representatives had received information from pharmacists and physicians that Contergan caused numerous side effects including abnormally cold hands and feet, paraesthesia and giddiness, Grünenthal replied to Voss saying “no such side effects have come to our notice” (10).
Nonetheless Voss described his three cases at a neurological congress in Düsseldorf on April 30th 1960. The presentation by Voss prompted numerous other reports of apparent severe peripheral neuritis in long-term users of Contergan. Frenkel, a neurologist from Königstein, who like Voss had contacted Grünenthal earlier with concerns about the safety of Contergan, submitted a paper to Medizinische Welt describing 20 patients he believed to have thalidomide-induced peripheral neuritis. Grünenthal representatives attempted to persuade Frenkel to withdraw or delay publication but he refused. However, for reasons that are unclear, Frenkel’s paper did not appear in print until May 6th, 1961. This was well after the first literature report describing thalidomide-induced peripheral neuritis which was from Dr A. Leslie Florence of Aberdeenshire, United Kingdom. In a letter to the British Medical Journal of December 31st 1960, entitled “Is thalidomide to blame,” Florence described 4 patients presenting with marked paraesthesia affecting first the feet then the hands, coldness of the extremities, ataxia and nocturnal cramp. All patients had been taking thalidomide for between eighteen months to two years. Cessation of the drug resulted in alleviation but not removal of the symptoms (11). By the end of May 1961 there were at least 1,300 cases of peripheral neuritis associated with long-term thalidomide therapy, and Grünenthal were forced to take steps to have the drug supplied only on prescription. This side action was soon to appear to be insignificant compared to the catastrophic result of ingestion of the drug by women during days 35-50 of their pregnancy, but the broad dissemination of the fact that thalidomide caused nerve damage was partially responsible for alerting Frances Kelsey, the FDA medical officer responsible for evaluating thalidomide before its use in the USA, to the fact that this supposed non-toxic medicine was potentially dangerous. The consequent delay in the approval of thalidomide until after its teratogenic effects were established in Europe thus averted a disaster for the United States (12).
Thalidomide and Fetal Abnormalities
The first indication of the emergence in Germany of an epidemic of particular fetal deformities was the description in 1959 by a Munich gynaecologist, Weidenbach, of a child with phocomelia of the arms and legs (13). Weidenbach knew this to be a highly unusual type of deformity since he could find no silmilar case in the German literature, nor had any case been described in the records of the Bavarian Institution for Crippled Children since its institution in 1913 (six years later, in 1965, four years after thalidomide had been withdrawn in Germany, Weidenbach finally established that the mother of the deformed child had been prescribed the thalidomide-containing preparation Grippex for a febrile condition between the 25th and 35th day post conception).
Other sporadic cases were reported but strong evidence that these fetal abnormalities had a common exogenous cause only emerged in September 1960, when Kosenow and Pfeiffer, at a meeting of the German Paediatric Association, presented details of two cases of children born with severe skeletal malformations together with various other deformities such as duodenal stenosis and capillary hemangioma of the upper lip. Of great significance, both children had been born on consecutive days (February 28th and 29th) in the same small German town. Some years later it was finally established that both mothers had been prescribed thalidomide between the 44th and 46th postmenstrual day (9).
By the beginning of 1961 it was clear that there was an epidemic of limb deformities in Germany, typically shortening or absence of the long bones of the arms or legs, producing flipper- or seal-like limbs, the condition designated “phocomelia.” Lenz was certain that one single common cause was responsible (14). He asked one affected parent specifically whether any new drugs had been taken during pregnancy. He received a detailed list of the diet and diseases of the mother and a list of ten drugs. Contergan was not mentioned.
By August 1961, Lenz estimated that the incidence of gross defects of the long bones had increased 10-fold in Hamburg and probably 100-fold or more in some towns of Rhineland and Westphalia. In September, 1961 Wiedeman published a paper in which he described seeing in his clinic in Krefeld only four cases of limb deformity between 1950-59, but 13 cases in the last 10 months (14). Careful investigation excluded infection, irradiation, anticonception chemicals and attempted abortion as possible causes, but Wiedeman was convinced that a newly introduced toxic factor was responsible.
Thalidomide (as Contergan) was originally considered as a candidate by the paediatrician Weicker, who in August 1961 had collected detailed data on 20 cases, and found Contergan was mentioned in five. Unfortunately, he ignored the possible connection since he was mistakenly informed that thalidomide was widely used in the United States, where no cases of phocomelia had occurred (12). In November 1961 Lenz visited two further cases and established that both mothers had taken Contergan during pregnancy. Lenz told Weicker of his suspicions and the latter rechecked his now considerable collection of case histories and found 34 cases with positive evidence of thalidomide use by the mother. Encouraged by this and other confirmatory evidence from Professors Wiedeman (Kiel) and Kosenow (Krefeld), Lenz telephoned Grünenthal describing his concern. He was told he would be visited by representatives of the firm within a few days. Lenz considered the problem too serious for delay and, on November 16th, 1961 sent to Grünenthal a registered express letter detailing his reasons for assuming a connection between use of Contergan in early pregnancy and consequent birth defects, particularly limb deformities (14).
In Australia, where thalidomide was marketed as Distaval by the Distillers Company, the connection between the drug and fetal abnormalities was deduced rather more rapidly through the perception of the obstetrician McBride. On May 4, 1961 at the Women’s Hospital, Sydney, McBride delivered a baby with bowel atresia and phocomelia of both arms. An obstetrician of 7 years standing, this was the first case of phocomelia McBride had seen at this hospital where there were 4,000 births each year. Twenty days later another child with similar limb malformations was delivered at the hospital. Two cases could be a coincidence, but when a third case with similar limb malformations was delivered on June 8th, 1961 McBride’s suspicions were acutely aroused. Over the following days he examined the literature on drug-induced malformations and consequently examined the case notes dealing with the three pregnancies; the only drug administered to the mothers was Distaval.
On June 13th, 1961 McBride persuaded the Medical Superintendent at the Women’s Hospital to withdraw thalidomide from use. McBride also attempted to initiate some animal experiments, firstly using mice and guinea pigs kept at the hospital for diagnostic purposes. Lack of sufficient animals and McBride’s understandable lack of expertise prompted him to approach, on two occasions, the professor of pharmacology at the University of Sydney to suggest he examine the possible teratogenic potential of thalidomide in laboratory animals. Unfortunately, the professor was not convinced by McBride’s circumstantial evidence and did not consider the expense of an animal study justified (15).
McBride informed representatives of Distillers in Australia of his suspicions that thalidomide might be responsible for producing fetal abnormalities, and was assured by them that his anxieties would be transmitted to the London office. McBride nonetheless drafted a letter to the prestigious medical journal The Lancet stating that although congenital abnormalities can be expected in approximately 1.5% of babies, he had observed that the incidence of multiple severe abnormalities in babies delivered of women who were given the drug thalidomide during pregnancy to be almost 20%. McBride’s letter, which appeared on December 16, 1961 was the first published record of the possible teratogenic action of thalidomide (16).
Distillers received the report of McBride’s concern from their Australian representatives on November 21, 1961. This, together with the co-incidental information from Lenz in Germany resulted in the withdrawal of thalidomide from the German and British markets. The incidence of thalidomide-induced deformity in Germany, together with a record of other significant observations is depicted in Figure 1.
With 50 years hindsight, some may view with incredulity the delay in the association of thalidomide with the ever increasing number of cases of severe congenital defects. Yet even in April 1962, Josef Warkany, then probably the most distinguished expert on teratogenic agents, expressed doubts that thalidomide was the cause of the epidemic of limb defects, since some mothers who had taken thalidomide during pregnancy produced normal offspring, and some mothers who gave birth to children with phocomelia claimed not to have taken the drug (17). These doubts disappeared when it was established that mothers who took thalidomide after the sensitive period of 50 days into pregnancy could have normal children; and mothers who did not believe they had taken the drug could have taken it unknowingly, since it was present in some preparations without being specifically named (18). As Warkany stated: “The real proof came when disappearance of the epidemic followed withdrawal of the drug from the market.” (18) (See Figure 18.1)
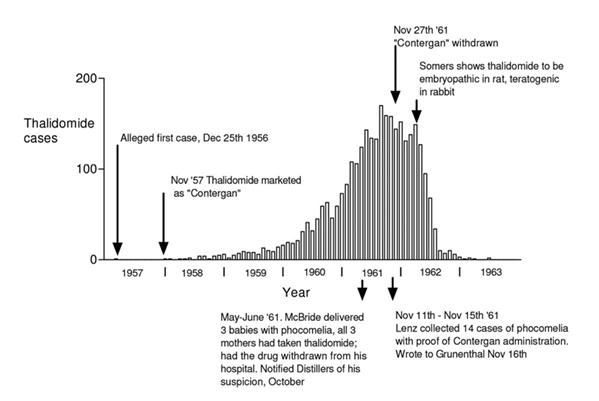
Fig. 18.1 Columns represent the monthly incidence in Germany of births of children deformed because of ingestion of thalidomide by the mother. Data was collected by the meticulous retrospective epidemiological studies of Widukind Lenz (5). The first case occurred in December, 1956. The child’s father worked for Chemie Grünenthal and had received samples of thalidomide tablets for his wife. The association of the abnormalities with administration of thalidomide was not suspected until May 1961 in Australia, and November 1961 in Germany. The rapid fall in incidence of deformity after withdrawal of thalidomide confirmed that thalidomide was responsible. In April 1962, 5 months after the withdrawal of the drug, Somers was the first to demonstrate the embryopathic action of thalidomide in animals.
Experimental Studies
Upon hearing of the concerns of Lenz and McBride, the chief pharmacologist of Distillers in Britain, Dr George Somers, launched studies of the effect of thalidomide on the fetus. Thus November 1961, after the withdrawal of the drug, was the first time thalidomide was administered to pregnant animals, apart from the unsophisticated attempts by McBride four months previously.
Somers published his results barely five months later, on 28 April 1962 also in a letter to The Lancet (1). In this letter he stated that thalidomide given to pregnant rabbits resulted in the birth of neonates in which: “The front legs were foreshortened owing to a reduction in long-bone formation of the radius and ulna; while the rear legs showed a varus deformity involving the tibiofibula.”
Therefore, within six months of the suggested association between thalidomide and human fetal deformity, it had been possible to produce in a common laboratory animal a teratogenic effect identical to one of the many produced by thalidomide in humans. Surely this was evidence for a striking similarity between mammalian species rather than for a manifest difference. Somers used a dose (150 mg/kg) that was higher than that used in humans. Somers simply wanted to know if thalidomide was teratogenic in rabbit and thus did not carry out a dose-response study, which would have been wasteful of animal life. However, he subsequently obtained the same effects with a 30 mg/kg dose as did Seller with 50 mg/kg (19). Somers also later measured plasma concentrations of thalidomide after oral administration of suspensions in carboxymethylcellulose and found that with doses of 150 mg/kg the peak plasma concentration in rabbits was only three-times as high as those in humans on normal doses (19).
Within a further six months, again in The Lancet, a report appeared describing the teratogenic effects of thalidomide in the Sprague Dawley rat (20) and this was confirmed by other reports (21). However, a more common response of the rat fetus to the challenge of thalidomide was intrauterine death followed by resorption. In the two years following the withdrawal of thalidomide approximately 20 papers were published that reported decreased litter sizes, together with numerous uterine resorption sites, in rats given the drug during pregnancy. Christie (22), who also reported a significant increase in resorptions in thalidomide-treated pregnant rats, suggested that fetal abnormalities produced in the rat embryos might well be of sufficient severity to kill the embryo and thus account for the increase in resorption.
Other papers subsequently appeared showing negative effects of thalidomide on the fetuses of laboratory animals. Obviously, when such conflicting results occur one assumes that the negative results are wrong, for there may be many reasons for a false negative, but a positive result is unlikely to be due to chance provided adequate controls are carried out. There were probably a number of factors responsible for the false negative results. Teratology was an orphan discipline in the 1960s, with few practitioners. Inexperienced experimentalists were possibly not aware that laboratory animals, which generally give birth at night, cannibalise their pups if they are born deformed, thus the litters should be delivered by Cesarean section at term to be sure of detecting deformed neonates. Also, detecting deformities (particularly of the skeleton) in fetuses of small laboratory animals such as rats and mice is not easy; the technique of staining with alizarin dye is necessary. King and Kendrick (20) for example, found that they could double the detection of skeletal defects in rat by alizarin staining of the skeleton.
The most significant reason for the false negative results was exposed by the definitive biochemical studies of thalidomide by the group led by Professor R.T. Williams at St Mary’s Hospital in London. Williams and his colleagues established that the distinct optical isomers of thalidomide were far more toxic than the mixture of the isomers (the racemate) (23). Subsequent studies of the relative effects of the separate enantiomers in the rat suggested that the teratogenic action was due to the S(-)-enantiomer. Since there is a rapid interconversion of the two isomers in vivo the role of stereochemical factors in thalidomide teratogenesis is probably significant but unresolved (24). Of greater significance, Williams et al. also showed that thalidomide was inherently unstable, rapidly breaking down to 12 inactive metabolites if in aqueous solution above pH 6 (25). It is probable that many workers were using solutions containing only the inactive metabolites of thalidomide and therefore obtained no fetal abnormalities. This is particularly likely, since in the British patent specification (1957) Grünenthal, commenting on the lack of solubility of thalidomide, stated quite erroneously, that thalidomide “is soluble in strong lyes, the solution obtained being yellowish in colour” (26). Such a treatment would result in the instantaneous breakdown of the substance. Chemical breakdown was certainly the reason for the apparent lack of teratogenic activity in rabbit reported by Fox et al. (27). Schumacher (28) repeated these experiments and showed that the procedures used by Fox to solubilise thalidomide resulted in considerable chemical breakdown, an observation subsequently accepted by Fox (29). Schumacher also emphasised the facilitating effect of carboxymethylcellulose (a suspending agent frequently used to make supersaturated solutions of thalidomide for experimental use) on hydrolysis. Carboxymethylcellulose contains from 7 to 8.5% sodium and therefore would tend to buffer the solution, maintaining the alkaline milieu that would foster the hydrolysis of thalidomide (28).
Unfortunately, many early papers on the experimental production of abnormalities in laboratory animals neglected to record how solutions or suspensions of thalidomide were prepared and for how long they were stored, but it is likely that negative results may well be explained by lack of absorption of the drug or the inadvertent administration of hydrolysis products rather than thalidomide itself. By 1967, Meredith Runner had stated that the embryos of all mammals so far studied are affected by thalidomide (30) and in 1989, Schardein and Keller listed 17 mammalian species in which thalidomide produced fetal abnormalities (2). It also produces limb deformities in the newt (31).
Thalidomide and the Regulation of Drug Use
The catastrophic effects of thalidomide on the developing fetus shook the confidence of the general public in the pharmaceutical industry. The dramatic improvement in the treament of infective diseases such as puerperal sepsis, pneumonia and streptococcal septicemia by the sulphonamides and penicillin, and the control of tuberculosis by streptomycin, isoniazid and aminosalicylic acid were all forgotten amid the realisation that there was no procedure in place that could prevent the marketing of a medicine that manifested a horrific toxic effect. As stated by the British Minister of Health, the Right Honourable Kenneth Robinson in May 1963: “The House and the public suddenly woke up to the fact that any drug manufacturer could market any product however inadequately tested, however dangerous, without having to satisfy any independent body as to its efficacy and safety and the public was almost uniquely unprotected in this respect.” (32)
To remedy this situation a joint subcommittee of the English and Scottish Standing Medical Advisory Committees was set up to advise the Government on suitable measures to ensure that potential medicines were subjected to intensive preclinical investigation to establish their pharmacological and possible toxic actions, and to confirm their efficacy and safety in the clinic. A further remit of the Subcommittee was to advise on procedures to ensure early detection of untoward effects that may emerge after the marketing of the product and its widespread use in patients. After discussions with the Association of the British Pharmaceutical Industry (ABPI) and other professional bodies the Subcommittee recommended that the responsibility for the preclinical investigation of new drugs should remain with the individual manufacturer and that there should be an expert body created that would review the evidence and offer advice on the toxicity of new drugs. Thus, in June, 1963 the Committee on the Safety of Drugs was established under the chairmanship of Professor Derrick Dunlop (later to become Sir Derrick). It became the responsibility of this committee to assess the data presented in support of each preparation and to consider whether it may be released for marketing. The assessment of the experimental studies on the toxicity of the drug was the responsibility of a subcommittee on toxicity (32). As a minimum requirement this subcommittee would expect the drug to have been tested according to the recommendations in the report prepared for the ABPI by the Expert Committee on Drug Toxicity (33). This report describes in detail the recommendations for the general investigation of the acute, subacute and chronic toxicity of a compound, its possible carcinogenicity and its liability to interact in a toxic fashion with concurrent medication. Not surprisingly, since the stimulus for the institution of regulations governing the marketing of drugs was the toxic action of thalidomide on the fetus, the report recommended particular tests to examine possible effects of the drug on fetal development.
The recommendations were that two species should be used to study toxic effects upon the fetus, the mouse or rat, and the rabbit. Test groups should be large enough to ensure 5 pregnant animals in the control rodent group and 3 in the rabbit. Three doses of the test drug should be used: the maximum dose tolerated by the mother, a second dose large enough to produce clinically relevant effects, and an intermediate dose. Dosing should begin on day one of pregnancy and continue until the day before term. On the day before term the animals should be killed, the uteri should be removed and examined for number of resorption sites, and the number and weight of live, dead or abnormal fetuses should be determined. From this preliminary test a range of doses should be selected to establish the threshold dose for fetal loss or any aspect of fetal toxicity. In this main test, the number and weight of progeny should be monitored up to weaning, when the progeny are killed and examined externally and internally. The normality of the skeleton should be observed after staining with alizarin red.
There is no doubt that if these premarketing procedures, instituted in the early 1960s, had been applied to thalidomide its toxicity to the developing embryo would have been detected. Unlike the situation in the United Kingdom, the United States did have some legislation to regulate the marketing of novel medications. New drug applications had to be submitted to the Food and Drug Administration (FDA) to be cleared for marketing, which at that time was on the basis of safety claims alone. The FDA had 60 days to decide if the safety data supplied was adequate. A failure to communicate by day 60 would mean automatic approval of the drug. An American pharmaceutical company signed an agreement with Grünenthal to market thalidomide in the United States and submitted an application for approval to the FDA on September 12 1960. According to Frances Kelsey, the medical officer assigned to the review of the thalidomide application, “Deficiencies in all areas were found during the initial review and in several subsequent resubmissions” (12). Kelsey was particularly concerned about the report of peripheral neuritis as a side effect, of which the FDA were only informed in February 1961, despite the fact that in Britain thalidomide had carried a warning of the risk of peripheral neuritis since September 1960. The severity of peripheral neuritis as a side effect led to questions of its use during pregnancy, since the application had described its use for the treatment of insomnia in pregnancy; yet there were no data as to whether it was safe to use in this condition (12). The legitimate concerns raised by Kelsey delayed the approval of thalidomide until its withdrawal from the market in Europe and Australia in November, 1961.
Even though the requirements of the FDA and Kelsey’s perception prevented the marketing of thalidomide in the United States, a few cases of thalidomide deformity did occur there since the company seeking approval to sell the drug distributed free samples of thalidomide tablets to over a thousand doctors who administered them to an estimated 20,000 patients. The realisation that potentially noxious medicines could be administered to patients prior to the approval by the FDA resulted in the Kefauver-Harris Amendment, which required that the FDA should monitor all stages of drug development before its use in humans (12).
Misrepresentation
An objective examination of published papers and contemporary accounts confirms that the preclinical tests upon thalidomide were cursory in the extreme, and there is no doubt that it was never administered to pregnant animals prior to its use in patients. Further, within a short time of thalidomide’s withdrawal from the market due to its suspected association with fetal abnormalities, it was shown to produce fetal toxicity in laboratory animals. Clearly, the disaster occurred because of insufficient testing in laboratory animals.
Antivivisection organisations are however loath to accept that the thalidomide disaster offers no propaganda for their cause, asserting in leaflets and newspaper advertisements that “The thalidomide disaster is just one example of how vivisection damages us.” (34) In the United Kingdom the Advertising Standards Authority (ASA) ensures that all advertisements should be “legal, decent, honest and truthful, and when capable of objective assessment, data should be supported by evidence.” Upon assessing the above statement the ASA ruled that: ”The common claim, that thalidomide was tested in various species and did not show the teratogenic effects that it had in humans, was considered unjustified.” The agency therefore requested that reference to thalidomide in this context in future advertising be omitted (35).
Unfortunately, there are no strictures on the perpetration of misrepresentations in general literature. One of the more bizarre descriptions of the history of thalidomide is that of Greek and Greek, who state that some toxicity tests were carried out on pregnant rodents prior to thalidomide’s release (36). This is not so. In fact one of the lines of defense used by the suppliers of the drug was that safety tests were not normally carried out on pregnant animals at the time thalidomide was developed. However, the preclinical investigation of meprobamate (described above) shows this claim to be false. Greek and Greek also state that it was known for five years that thalidomide was teratogenic in humans but “since animal testing had not indicated a problem with thalidomide, its use persisted. Hence animal testing delayed the recall of this highly teratogenic drug.” The reviews by Lenz, Warkany and Kelsey, the leading protagonists in this field, published as the proceedings of a symposium to commemorate the 25th anniversary of the American Society of Teratology (5, 12, 17), show that the account by Greek and Greek is wildly inaccurate. However such propaganda has been eagerly grasped by antivivisectionists and widely quoted in letters to the press and in internet discussions. The public must be constantly reminded of the facts before they are swamped by the erroneous, emotive rhetoric of the opponents of animal experiments.
An earlier version of this chapter was published as: Botting, J. The history of thalidomide. Drug News and Perspectives 2002, 15(9): 604-11. Copyright 2002-2014 Prous Science, S.A.U. or its licensors. All rights reserved, http://dx.doi.org/10.1358/dnp.2002.15.9.840066
References
- Somers, G F (1962) Thalidomide and congenital abnormalities. The Lancet 1 912.
- Schardein, J L and Keller, K A (1989) Potential of human developmental toxicants and the role of animal testing in their identification and characterisation. CRC Crit. Rev. Toxicol. 19 251-330.
- Kunz, K, Keller, H and Mückter H (1956) N-Phthalyl-glutaminsäure-imid. Arzneim.-Forsch. 6 426-30.
- Jung, H (1956) Klinische Erfahrungen mit ein neuen Sedativen. Arzneim.-Forsch. 6 430-32.
- Lenz, W (1988) A short history of thalidomide embryopathy. Teratology 38 203-15.
- Somers, G F (1960) Pharmacological properties of thalidomide (α-phthalimido glutarimide), a new sedative hypnotic drug. Brit. J. Pharmacol. 15 111-16.
- Sunday Times of London (1979). Suffer the Children: The Story of Thalidomide. London: Andre Deutsch, p. 59.
- Berger, F M (1954) The pharmacological properties of 2 methyl-2-N-propyl, 3 propanediol dicarbamate (Miltown), a new interneuronal blocking agent. J. Pharmacol.Exp. Ther. 112 413-23.
- Lenz, W (1979) Thalidomide: facts and inferences, in Drug-Induced Sufferings. Int. Congr. Ser. No. 513. Proc. Kyoto Int Conf Against Drug-Induced Sufferings, pp. 103-09.
- As Ref. 7. p. 32.
- Florence, A L (1960) Is thalidomide to blame? Brit. Med. J. 2 1954.
- Kelsey, F O (1988) Thalidomide update: regulatory aspects. Teratology 38221-25.
- McBride, W G (1977) Thalidomide embryopathy. Teratology 16 79-82.
- Lenz, W (1985) Thalidomide Embryopathy in Germany, 1959-1961. Prevention of Physical and Mental Congenital Defects, Part C: Basic and Medical Science, Education, and Future Strategies, pp. 77-83.
- As Ref. 7. pp. 86-95.
- McBride, W G (1961) Thalidomide and congenital abnormalities. The Lancet 2: 1358.
- Warkany, J (1988) Why I doubted that thalidomide was the cause of the epidemic of limb defects of 1959 to 1961. Teratology 38 217-19.
- Lenz, W (1965) Epidemiology of congenital malformations. Ann. NY Acad. Sci. 123 228-36.
- Somers, G F (1963) The foetal toxicity of thalidomide. Proc. European Soc. Study Drug Toxicity 1 49.
- King, C T G and Kendrick, F J (1962) Teratogenic effects of thalidomide in the Sprague Dawley rat. The Lancet 2 1116.
- McColl, J D, Globus, M and Robinson, S (1965) Effect of some therapeutic agents on the developing rat fetus. Toxicol. appl. Pharmacol. 7 409-17.
- Christie, G A (1962) Thalidomide and congenital abnormalities. The Lancet 2 249.
- Fabro, S, Smith, R L and Williams, R T (1967) Toxicity and teratogenicity of optical isomers of thalidomide. Nature 215 296.
- Shah, R R, Midgeley, J M and Branch, S K (1998) Stereochemical origin of some clinically significant drug safety concerns: lessons for future drug development. Adverse Drug React. Toxicol Rev. 17 145-90.
- Schumacher, H, Smith, R L and Williams, R T (1965) The metabolism of thalidomide: The spontaneous hydrolysis of thalidomide in solution. Brit. J. Pharmacol. 25 324-27.
- Thalidomide Patent 1957. British Patent 768,821.
- Fox, R R, Sawin, P B, Crary, D D and Wuest, H M (1966) Intravenous injection of thalidomide in pregnant rabbits. Science 153 310.
- Schumacher, H, Blake, D and Gillette, J (1966) Thalidomide solutions. Science 154 1362.
- Wuest, H M and Fox, R R (1966) Thalidomide solutions. Science 154 1362.
- Runner, M N (1967) Comparative pharmacology in relation to teratogenesis. Fed. Proc. 26 1131-36.
- Bazzoli, A S, Manson, J, Scott, W J and Wilson, J G (1977) The effects of thalidomide on the generating forelimb of the newt. J. Embryol. Exp. Morphol. 41 125-35.
- Shah, R R (2001) Thalidomide, drug safety and early drug regulation in the U.K. Adv. Drug React. Toxicol. Rev. 20 199-255.
- Association of the British Pharmaceutical Industry (1964) First Report of the Expert Committee on Drug Toxicity together with further Recommendations on Toxicity Evaluation.
- 1992. Press advertisement in the U.K. by the Antivivisection Agency.
- Advertising Standards Authority Report, Ref. B92-02904.
- Greek, C R and Greek, J S (2000) Sacred Cows and Golden Geese. New York: Continuum.

