
17. Genetic Trees, Admixture,
and Mosaics
© 2024 Marianne Sommer, CC BY-NC-ND 4.0 https://doi.org/10.11647/OBP.0396.21
We have seen in the last chapter that Cavalli-Sforza’s and colleagues’ human population genetics was centrally about tree building. From the beginning, the assumptions underlying this approach were also criticized. There was some debate about what constituted the superior kind of data. Howells (1973b), for example, initially held that the genetic tree did not stand for true phylogeny. In his view, serological traits were not the best characteristics for studying population histories, but Howells’ critique was not a general one against tree building. To the contrary, as we have seen, he was a tree builder, and also in this paper he included a tree based on cranial measurements (175). Other (physical) anthropologists issued more fundamental criticism, however. Ashley Montagu, C. Loring Brace, and Frank Livingstone denied the very existence of human genetic populations. Livingstone used the concept of the cline (introduced by Julian Huxley [1938b]) to describe the fact that a trait varies continuously not abruptly across space. Furthermore, the frequencies of different alleles did not fall together to form clusters (e.g., Livingstone 1962; see also my discussion of Huxley and Morant in Part III). The tree model was therefore not accurate, and Livingstone (1991) was among those who would demonstrate that the correlation between genetic and geographic distance that underlay tree building could be accounted for by other models than binary fission, such as genetic exchange between relatively stable neighboring populations (similar to the isolation by distance model).1
Another cautioning voice along these lines was the physical anthropologist Gabriel Lasker (1976) who pointed to the particularly difficult situation for human population studies, due to the influences of parallel evolution, culture, and interbreeding. The biochemical methods for studying primate and intra-human phylogenies were essentially the same, but in the latter case the branches of a cladogram could not be read as representing reproductively isolated groups. Such a cladogram could at best be approximate, since the branches in a phylogenetic tree that represented human variants were in reality interconnected. The British-born American statistician, Elizabeth A. Thompson (1975), too, observed that human populations often did not fulfil the criterion of being isolated, non-interbreeding, required by the tree schema, even while her in-depth treatment of the theoretical, evidential, and computational aspects of population-genetic tree building supported the notion that the available genetic data did not warrant more sophisticated models. Thus, also in controversies within the young field of human population genetics itself, the question of whether trees were an adequate representation, or rather gave a false impression of evolutionary history because of degrees of interbreeding, was certainly an issue (see, e.g., also, Kirk 1969; Lalouel 1974; Morton 1974; Cavalli-Sforza 1974).
We have seen that Cavalli-Sforza and colleagues, too, to a certain extent recognized the limits of the tree shape and showed an interest in questions of admixture early on. From the outset, Cavalli-Sforza actually suggested that the tree shape might only work for populations that are geographically far apart, because otherwise “[i]nstead of a ‘tree’ one may have to estimate a ‘network’; such methods do not yet exist” (Cavalli-Sforza 1973, 96; Sommer 2015a, 120–21). With regard to “such methods”, the geneticist Ranajit Chakraborty published a review in 1986. Under the premise that “[i]n humans, exchange of genes between populations separated by large geographic distance and wide cultural and/or political barriers have [sic] been in operation since millennia” (1), he discussed the history and state of research on admixture, beginning with Felix Bernstein’s studies on the distribution of blood groups published in 1931.
Bernstein, who had worked on the inheritance of blood groups with statistical methods, directed the Institute of Mathematical Statistics at the University of Göttingen (Germany) and lectured on biomathematics. In the 1931 publication, he presented his attempts at reconstructing the migrations and mixtures of peoples on the basis of current blood group frequencies, such as the dispersal of the B-gene in relatively recent times from central Asia. Bernstein’s considerations in general led him to argue against the polyphyletic views of human evolution and kinship that existed at his time and that I have discussed in Part III. He insisted that, during their entire existence, human groups had been part of processes of mixture as well as diversification. Thus, he rejected the image of the tree also for a monophyletic understanding of the origin and kinship of “Menschenrassen” [human races] (17), even while retaining a vegetal metaphor:
The family tree of humanity does therefore neither resemble the image of a tree, nor the image of several trees grown together, which stem from separated roots, but the intergrowth and intertwining is so manifold, already at the roots, that we must view each putatively pure stem as a mixture [even] with regard to certain very old characteristics. (Bernstein 1931, 19, my translation)2
By the time Chakraborty published his review on admixture research in the mid-1980s, there were different methods in use to estimate the relative contributions of ancestral populations to a new hybrid population, some of which were applicable to two ancestral populations, while others allowed for more than two. The models therefore assumed that two or more existing populations gave rise to a new, hybrid population. African Americans were the most studied ‘population’, followed by the interest in (the Nordic admixture in) Icelanders and (the gentile admixture in) Jews. For over a decade, admixture had also gained attention on the level of the individual (proportions of ancestry for a hybrid individual), again with a special focus on African Americans.3 Long before commercial ancestry tracing companies would pop up, studies even began to suggest the possibility under certain conditions to attribute specific genotypes to ‘their populations’ (Spielman and Smouse 1976; Smouse, Spielman, and Park 1982).
These strands of research were seen to be in their infancy, and it was hoped that the availability of many DNA sequences would help resolve some of the problems the study of admixtures and times of divergence with classical markers had so far encountered. For the future of admixture research discussed below, it is relevant that it was, on the one hand, assumed that “it may not be far from reality to conclude that admixture of different ethnic groups during the evolutionary history of man has resulted in some degree of homogeneity of genetic variation among populations” (Chakraborty 1986, 35). On the other hand, there also surfaced the notion of originally pure populations. The research demanded precise knowledge of the allele frequencies in all populations in a study. This was seen as a challenge as parent populations may no longer be available “in their original, unaltered form”, i.e., “in an unmixed state” (Chakraborty 1986, 21; see also Thompson 1975, 134). Also relevant to the following is Chakraborty’s observation that not enough attention was being paid to the historical hypotheses behind the research: “In human populations, admixture generally does not occur with a single sudden influx. The process of admixture in most admixed human groups had been more like the ebb and flow of tidal waves […]” (1986, 9).
Building on a paper Cavalli-Sforza and Piazza had published in 1975 that mostly defended tree building, in The History and Geography of Human Genes, Cavalli-Sforza, Menozzi, and Piazza (1994, 54–9) included a section on admixtures, their estimations and (distorting) effects on tree structures. They mathematically described how to approximate the time that had elapsed between the separation of the ancestral populations and their admixture. They discussed the calculation of the respective percentages of the contributions of ancestral populations to an admixed population (for the case of African Americans). They also introduced an artificially admixed population (a population created from 60%-English and 40%-Ainu ancestry) into the estimation of a population tree to discuss the effects. Finally, the treatment of investigations of differences in autosomal DNA sequences led them to speculate that Europeans resulted from an admixture between Chinese and African populations (with the latter contributing less) – a hypothesis they visualized by drawing on a paper Cavalli-Sforza had co-signed (Bowcock et al. 1991, 168–71) (see Figure IV.9).
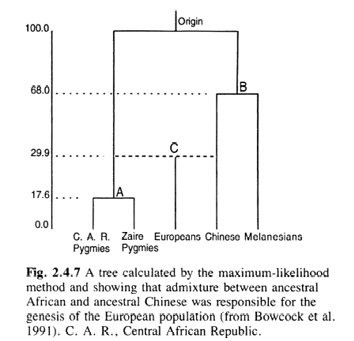
Fig. IV.9 A tentative breach of the human family tree: Europeans as ‘Chinese-African admixture’ (all rights reserved; used with permission of Princeton University Press, from Luigi Luca Cavalli-Sforza, Paolo Menozzi, and Alberto Piazza, The History and Geography of Human Genes [Princeton: Princeton University Press 1994], Fig. 2.4.7,
p. 92; permission conveyed through Copyright Clearance Center, Inc.).
However, Cavalli-Sforza, Menozzi, and Piazza mainly pointed to the problems with regard to admixture studies, especially where the admixture lay deep in the past and the ancestral populations were (genetically) unknown. Thus, they still concluded that although
“[i]n theory it is possible to construct a tree with connections between the branches […]” (1994, 58), in practice, geneticists had so far failed to reconstruct “true networks” (ibid., referring to Lathrop 1982). Generating trees with interconnections required an enormous amount of data. And even such interconnections would be shy of the likely course of history, since “[o]rdinarily population mixtures do not occur in a ‘catastrophic’ fashion, but are more likely to take place by the continuous slow infusion of individuals […]” (Cavalli-Sforza, Menozzi, and Piazza 1994, 55). In sum, “[t]he full analysis of reticulate evolution remains an important task for the future” (59). Until then, one might exclude populations suspected of admixture from tree building. In The History and Geography of Human Genes, the Cavalli-Sforza and Edwards (1965) genetic tree of human populations and its projection on a map in Edwards and Cavalli-Sforza (1964) were therefore reproduced with the remark that they assumed independent evolution in the branches of the tree, meaning “no important fusions or exchanges between the branches” (Cavalli-Sforza, Menozzi, and Piazza 1994, 69).
In other words, despite the assumption of admixture (fusions) and gene flow (exchanges), human relatedness was persistently forced into tree shapes; not in a genealogical way, however. In contrast to the genealogical family tree that links individuals on the basis of direct descent, these genetic population trees distance populations on the basis of overall genetic difference. The methods in human population genetics that are more analogous to genealogy were only just on the horizon, as indicated by the mitochondrial trees treated in Chapter 16 that actually (re)constructed the ‘descent’ of current DNA sequences in the sense of steps of mutations (‘maternal lines’). So did studies of polymorphisms in Y-chromosomal DNA (‘paternal lines’), the possibilities of which, too, were explored in The History and Geography of Human Genes. And in the context of autosomal DNA studies, Cavalli-Sforza, Menozzi, and Piazza expressed the wish that “[o]ne would like to be able to extend this approach including fusions as well as fissions in evolutionary human history, but accurate analysis of a greater number of populations would most probably demand information on many more genes than are available” (1994, 93).
This ability to extend the approach was not so long in the making. By the beginning of the third millennium, new statistical and computational approaches could be brought to bear on the analysis of an unprecedented amount of human genomic data. Expanding on the possibilities of clustering methods such as principal component analysis, statistical software like STRUCTURE (Pritchard, Stephens, and Donnelly 2000), FRAPPE (Tang et al. 2005), and ADMIXTURE (Alexander, Novembre, and Lange 2009) made it possible to group genetic samples into clusters and analyze the degree to which present-day individuals and populations are the result of genetic mixtures. With the introduction of programs for the graphical display of population structures like DISTRUCT (Rosenberg 2004), the visual black box of these seemingly discrete and homogenous entities – human populations – was opened. Individuals and populations came to be represented as colored bar plots indicating their admixed histories. Accordingly, the individual human genome as well as human diversity as such were now widely conceived of as a mosaic. A shift that, as we will see, eventually also registered in popularizations and commercialization.
Software like STRUCTURE allows the allocation of N individuals belonging to n geographically and/or ethnically defined populations to K groups so that these groups have the smallest within-population variation and the highest between-population variance. Starting with K equals 2, the method distributes the N individuals among just two groups, but also graphically visualizes the degree of admixture. This can be repeated with 3 K and so forth. The optimal number of K for the data is estimated in the process and depends on N and n. At the same time, there is a certain tendency to correlate K with conventional geographic regions, with the result that despite its quality of literally bringing to light the intermixed state of individual genomes and the admixed nature of populations, the clustering of individual genomes into populations seems to reify the age-old notion of ‘continental races’. This is in fact what happened in a genome-wide and global study with FRAPPE of which Cavalli-Sforza, aged eighty-six, was still a signatory (Li et al. 2008). In this “most comprehensive characterization to date of human genetic variation” (1100), the data of 938 individual genomes from fifty-one populations from the Human Genome Diversity Panel were said to segregate into the five continental groups. In the plotted results for K 7 with the DISTRUCT program, the seven clusters built with FRAPPE on the basis of the individual samples were labeled ‘Africa’, ‘Middle East’, ‘Europe’, ‘Central and South Asia’, ‘East Asia’, ‘Oceania’, and ‘America’.
Nonetheless, when experimenting with such programs, it seems that the more fine-grained the analysis becomes, the obscurer the populational structure gets. In other words: the bigger K, the more ‘previously pure populations appear as admixed’. This can be indicated by means of an online blogpost by Dienekes Pontikos, on which there is an ADMIXTURE analysis that moves from K 1 up to K 15. Figure IV.10 represents the analysis at K 15. The 15 clusters (K) are not separated by lines in the way usual for such visualizations, but only the 139 smaller populations that were studied and to which the 2,230 individuals whose DNA was analyzed belong (‘sample populations’). It is indeed a beautiful mosaic that evokes a cheerful picture of humankind in all colors of the rainbow.
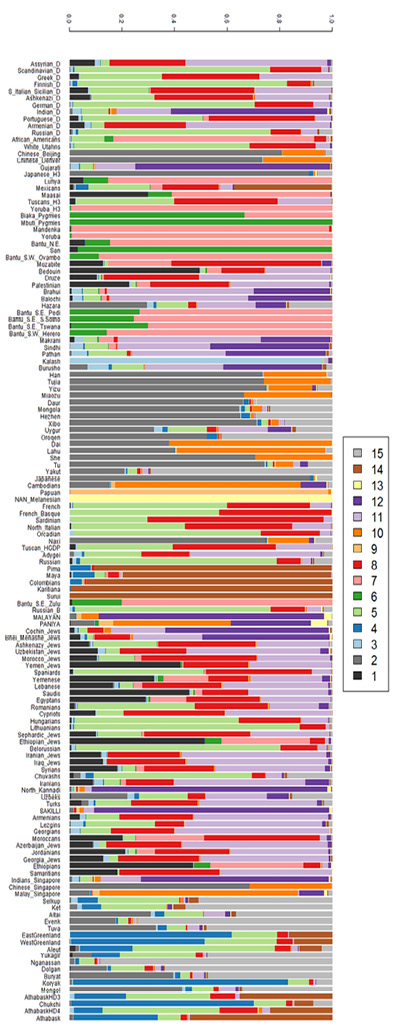
Fig. IV.10 Cluster diagram with K=15 (Dienekes Pontikos, “Human Genetic Variation: The First ? Components,” Dienekes’ Anthropology Blog [15 December 2010], http://dienekes.blogspot.com/2010/12/human-genetic-variation-first.html).
This tendency of ‘bursting individuals and populations’ seems to stand in stark contrast to the tree structure, which can be shown by Figure IV.11. It is a representation of the same data in the shape of a phylogenetic tree for the fifteen “original/ancestral components or populations” inferred by ADMIXTURE (the 15 K). In this process, the admixture disappears, and we return to a diagram that creates a hierarchical order from “Sub-Saharan” to “Siberian”. Instead of a human mosaic, in Figure IV.11 we again see the diaspora, in which populations (in this case fifteen) seem to have differentiated from a common source without converging (see also Sommer 2015a, 134–35; 2016a, 380–83).
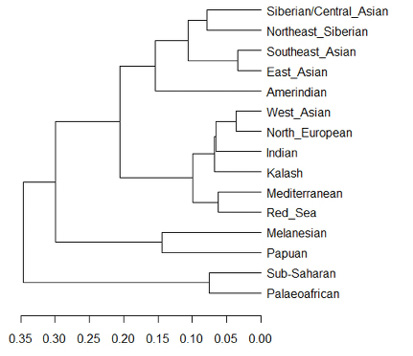
Fig. IV.11 Dendrogram of hierarchical clustering of the 15 ancestral components (Dienekes Pontikos, “Human Genetic Variation: The First ? Components,” Dienekes’ Anthropology Blog [15 December 2010], http://dienekes.blogspot.com/2010/12/human-genetic-variation-first.html).
However, let us take a closer look at the mosaic of Figure IV.10. The ‘ancestral populations’ (the 15 K) that are so neatly separated in the tree of Figure IV.11 are assumed also in this diagram. Even though the current populations and individuals are shown to form mixtures, they form mixtures of these supposed ‘pure ancestral populations’ that are marked by the fifteen distinct colors. Thus, the mosaic as well as the tree suggest a genetic order that existed before the major population movements took place. It seems that while admixture has become the center of attention in human population genomics, underneath its colorful diagrams still lurks the conception of originally pure populations hierarchically arranged in a tree – the origination in one population with successive distribution across the globe through fission without fusion. Thus, through altering the number of K, researchers may aim at exposing (tree-like) hierarchical relations between human populations. But assumptions built into STRUCTURE, ADMIXTURE, and similar programs, as well as the number of K assumed, lack rigorous statistical tests, and results may be interpreted subjectively, including as evidencing the existence of traditional ‘racial’ categories.4
Yet, visualizations of ADMIXTURE analyses and similar programs do (at least) show living individuals and current populations as considerably admixed. As a consequence, besides the tree (on a map) visualizations (as shown in Figure IV.8), diagrams that construct humans as genomically of mixed ancestries have also become current in popular and commercial contexts as is indicated by Figure IV.12 (even if with the simultaneous suggestion, through the distinct colors on the map and in the list, that the admixture has been between different, in themselves homogenous, individuals from pure populations).
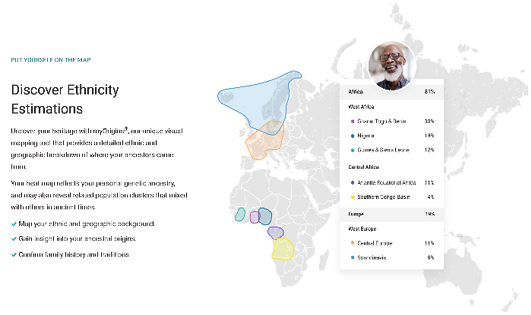
Fig. IV.12 Screenshot taken from a website of the genetic ancestry testing company Family Tree DNA (https://www.familytreedna.com/products/family-finder, last accessed 17 July 2023, with kind permission from FamilyTreeDNA).
In stark contrast, trees do not make admixture obvious, even though, besides purportedly giving an image of human relatedness before major intermixtures took place, they at the same time stand for relations between currently living human groups. The owner of the online blog and producer of the diagrams shown as Figures IV.10 and IV.11 is aware of the simplification a visualization as tree encompasses, but, reminiscent of Cavalli-Sforza’s approach, he still carries it out.5 The folding of time, this simultaneity of the non-simultaneous, that is inherent in such genomic trees is further evidenced by the fact that the ancestral population called “Palaeoafrican” in the tree of Figure IV.11 refers to the “Pygmies and San” (!), or rather to genomic data gained from individual human beings belonging to groups who have been given these names by outsiders. And this at a time, as will be of concern in Chapter 18, when aDNA has become part of a new field of enquiry that no longer reconstructs the deeper evolutionary history of modern humans on the basis of current genetic diversity alone (Sommer 2022b, 290–93; Sommer and Amstutz 2024, “Enter Ancient DNA: Mosaic and Trees”).
Before moving on to studies that include aDNA in the next two chapters, let us recall the view dominant at that time of the evolution of modern humans and the role archaic humans played therein. In The History and Geography of Human Genes (1994), Cavalli-Sforza, Menozzi, and Piazza worked with the tree shape of modern human evolution in support of the out-of-Africa model that assumed no interbreeding with archaic humans outside of Africa. They explicitly rejected the multiregional model of Franz Weidenreich, to the followers of which they, too, mistakenly counted Coon. The misunderstanding seems not to have ended there, because they claimed that the multiregional model assumes parallel evolution of the ‘racial’ lines in the different parts of the world, whereas, as we have seen in Part III, Weidenreich postulated genetic exchange between the regions and basically one evolutionary line or a network, rather than many parallel lines. Cavalli-Sforza, Menozzi, and Piazza (1994, 62–64) maintained this misrepresentation in word as well as diagram (see Figure IV.13).
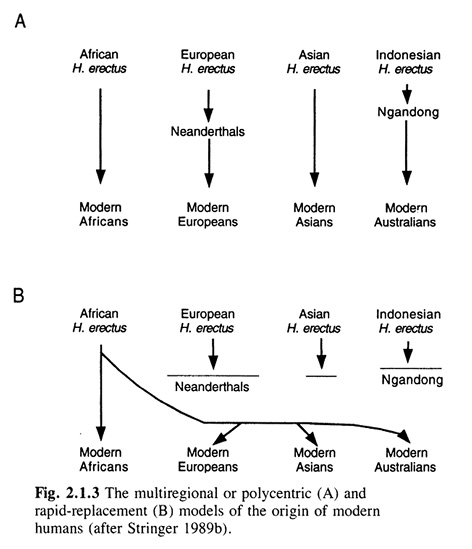
Fig. IV.13 Weidenreich’s model distorted to parallel evolutionary lines (A) and juxtaposed to the out-of-Africa replacement model (B) (all rights reserved; used with permission of Princeton University Press, from Luigi Luca Cavalli-Sforza, Paolo Menozzi, and Alberto Piazza, The History and Geography of Human Genes [Princeton: Princeton University Press 1994], Fig. 2.1.3, p. 62; permission conveyed through Copyright Clearance Center, Inc.).
The out-of-Africa model, which, when translated into a diagram, renders a tree for modern human evolution like in Figure IV.13B, stood for the notion that modern humans expanded “from Africa to Asia and the rest of the world, rapidly replacing the earlier human types living in these other regions” (Cavalli-Sforza, Menozzi, and Piazza 1994, 63). Although in their treatment of the archeological and paleontological knowledge, Cavalli-Sforza, Menozzi, and Piazza did not completely rule out a little genetic exchange, especially in east Asia, they maintained that “Cro-Magnon seems to emerge essentially unmixed” (65) and stated that “[i]f we look at the two hypotheses shown in figure 2.1.3 [Figure IV.13 above], we conclude with a definitive preference for replacement” (66).
In the next chapter, I engage with the developments taking place with the inclusion of aDNA data. This discussion is set against the backdrop of the preceding chapters, which highlighted the conundrum that while admixture between modern human populations has always been granted to some extent and gene flow between modern and archaic populations has not entirely been ruled out, the prevailing focus has been on building trees. Did the possibilities of including aDNA data in the analyses lead to novel ways of modelling human relatedness? This question again demands reflection on terminology at the outset. In The History and Geography of Human Genes, Cavalli-Sforza, Menozzi, and Piazza stated that “interconnected trees are networks. In the language of graph theory, trees bifurcate or multifurcate, but their branches do not connect” (1994, 58). However, in the following chapters we will see that there exist different notions of what a tree constitutes, and Chapter 18 will show that trees with a few connecting branches still look very much like trees.
1 Isolation by distance is a special case of gene flow. In this case, genetic exchange mostly takes place between neighboring populations, but genes can also spread to distant populations over many generations using intermediate populations as steppingstones (if there are no absolute barriers between them).
2 “Der Stammbaum des Menschen gleicht deshalb nicht dem Bilde eines Baumes, und auch nicht dem Bilde mehrerer miteinander verwachsener Bäume, die aus getrennten Wurzeln kommen, sondern die Verwachsung und Verflechtung ist eine so vielfältige, bereits von den Wurzeln her, dass wir jeden angeblich reinen Stamm in Bezug auf gewisse sehr alte Eigenschaften als eine Mischung anzusehen haben.” Under national socialism, Bernstein lost his position and temporarily emigrated to the US.
3 The medical geneticists Charles J. MacLean and Peter L. Workman positioned their work among other things in the interest in the genetic differences between “races” in gene frequencies with respect to behavioral traits such as IQ (1973a; 1973b, 341).
4 Programs such as STRUCTURE and ADMIXTURE have been criticized for diverse statistical problems, for the lack of a statistically rigorous justification for the number of K, as well as for the lack of tests for whether the K populations are genetically differentiated to a statistically significantly amount (Alan Templeton, personal communication, 8 January 2024; for a critical discussion of the program, see also Bolnick 2008; on different Bayesian algorithm models that do and do not take into account admixture and geographic information in the determination of population structure, see François and Durand 2010; for an alternative, network approach that does not seem to suggest ‘pure’ ancestral populations, see Greenbaum et al. 2019).
5 Dienekes Pontikos, “Human Genetic Variation: The First ? Components,” Dienekes’ Anthropology Blog (15 December 2010), http://dienekes.blogspot.com/2010/12/human-genetic-variation-first.html