
20. Deconstructing the Tree Diagram to a Mess – or at least
a Net
© 2024 Marianne Sommer, CC BY-NC-ND 4.0 https://doi.org/10.11647/OBP.0396.24
Even [tough] our brains search for a simple origin story, what we find is a beautiful mess. (Gokcumen 2020, 69)
The Linnean system is hierarchical and assumes clear-bounded entities or taxa. However, there are scientists, among them evolutionary anthropologists Isabelle C. Winder and Nick P. Winder (2014), who have argued that the tree-like Linnean order is still too strongly taken to be the norm, with mechanisms of reticulation, or heterarchical evolution, viewed as the exception. In fact, reticulation appears in many species of plants, insects, and mammals, including the primates, where it most often (but not exclusively) happens at the subspecies and species level. Thus, Winder and Winder (2014) have demanded a rethinking of the models of hominin evolution. Morphological and genetic data supported the notion that the species boundaries between hominins in Pleistocene Eurasia had been open and that genetic exchange across these boundaries had taken place. The mosaic appearance of traits in the Plio-Pleistocene fossil hominins suggested “a reticulating lineage exploring a complex, multi-dimensional space of possible morphologies and repeatedly generating and re-generating new combinations of traits” (306). The intra-specific structure of today’s Homo sapiens might be similar to the structure of reticulations at higher taxonomic levels in other primates and to the structure that once existed between different hominin species.
These observations point in the direction of a possible paradigm shift, wherein different questions may be asked, such as: in view of gene flow between species, is the biological species concept still meaningful? And how much gene flow must there be to finally undermine the hierarchical Linnean model, including the systematic and phylogenetic tree? Different diagrams are associated with these competing models. The first model of reticulate evolution in Figure IV.22 no longer has a tree structure, and the taxonomic system it suggests is not purely hierarchical but cross-cutting like Winder’s and Winder’s interpretation of the mosaic of characters in the fossil record of the hominin lineage.
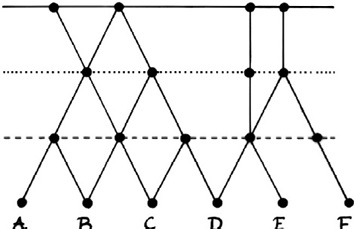
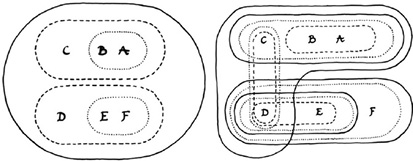
Fig. IV.22 Reticulate evolution and its cross-cutting taxonomy (top and bottom right) as opposed to the hierarchical taxonomy suggested by a tree-like phylogeny (bottom left). Isabelle C. Winder and Nick P. Winder, “Reticulate Evolution and the Human Past: An Anthropological Perspective” (Annals of Human Biology 41.4 [2014]: 300–311), Figs. 4 and 5, p. 308. All rights reserved, reprinted by permission of the publisher (© Taylor & Francis Ltd, http://www.tandfonline.com).
Similarly, the molecular phylogeneticist and bioinformatician David A. Morrison has criticized that “if we approach phylogenetics from the tree perspective then our only choice is to consider reticulations as additional (and unusual) occurrences” (2014a, 632). In fact, he found reticulation often to be “‘the last resort’” (633), the last possible evolutionary explanation that is taken into consideration. Conversely, Morrison described trees as a subset of networks in the sense that a tree is a network without reticulations. In his perspective, all phylogenies are networks, some of which are more tree-like than others. If a tree is a simplified network – as the genomic methods may suggest – “all trees are networks but not all networks are trees” (Morrison 2016, 457; also, 2014b). Most importantly, in phylogenetics, “[a] ‘tree (possibly with reticulations)’ is a less useful idea than a ‘network (which will be more or less tree-like)’” (Morrison 2014a, 635). Analyses should thus not start from a tree and add some reticulations, but from a network.
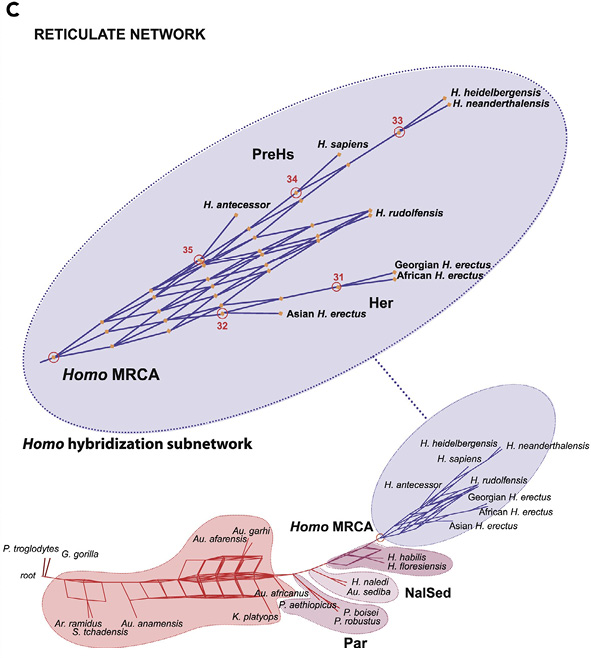
Fig. IV.23 “Homo hybridization subnetwork”. Miguel Caparros and Sandrine Prat, “A Phylogenetic Networks Perspective on Reticulate Human Evolution” (iScience 24.4 [2021]: 1–31), Fig. 4, p. 10, https://doi.org/10.1016/j.isci.2021.102359 © The Authors.
There are two groups of models: evolutionary history networks versus data presentation networks. The first entail hypotheses about evolutionary history, while the second simply describe the data, i.e., what complexities there are, which parts of the data contradict others, etc. With the second method, if the latter is the case, one has to figure out whether the contradictions are due to mistakes or whether different genetic histories are mixed together in the same organisms. Morrison warns that the two approaches may arrive at the same model, but one should not confuse the methods. He anticipates that in the future, biologists will start their investigation of data with the underlying complexity and work out the patterns from there, rather than setting out with assumptions regarding evolutionary history, and particularly not with the one that it is simple (personal interview with David Morrison, 7 November 2023).1
Contextualizing their paper in such network perspectives as taken by Isabelle C. Winder, Nick P. Winder, and Morrison (whom they cite), archeologist Miguel Caparros and paleoanthropologist Sandrine Prat (2021) combined a Maximum Parsimony and Phylogenetic Networks method (SplitsTree software) in the analysis of phenotypic craniodental features of twenty-two hominin species. They first arrived at a consensus tree, out of the conflicting trees suggested by different aspects of the data, which means a loss of phylogenetic information. To get access to this lost information, they proceeded to construct a consensus network and reticulate network and concluded that reticulation was a more informative framework to explain the relationships between the species of Homo.2 Figure IV.23 represents their “Homo hybridization subnetwork” from the reticulate network (Caparros and Prat 2021, Fig. 4, 10).3
Analyses similar to the one conducted by Caparros and Prat, focusing on features pertaining to the cranium or teeth, are carried out on the basis of genetic data. Researchers may produce a series of trees for different genome regions and then try to combine them into one tree of human populations. This is because different regions of the genome have different evolutionary histories. Indeed, the current understanding of a genome undermines everyday notions of genealogy and identity. In contrast to mtDNA that is handed down ‘intact’ from mothers to children, or the Y chromosome that is transmitted solely from fathers to sons, the nuclear DNA is freshly amalgamated between mother and father each generation, when both egg and sperm contribute one set of chromosomes. Beyond that, the chromosomes are regularly broken up and put together in new ways, thereby combining two different genealogical lines (that of the mother and father) on one chromosome, which will be passed on in this state to the next generation. In other words, different chromosomal fragments (so-called haplotype blocks) have their own ancestry or genealogy, rendering genomes – like the fossil record – mosaic in nature. Thus, individual chromosomal segments will ‘tell’ their own histories (e.g., Reich 2019, 10–16).
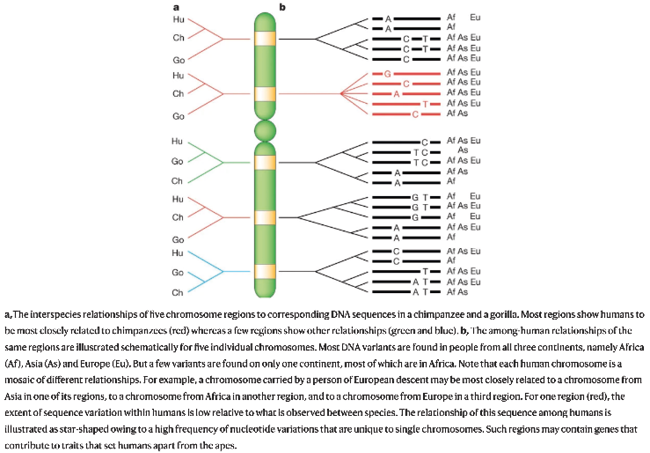
Fig. IV.24 The human genome “as a mosaic of haplotype blocks”. Svante Pääbo, “The Mosaic That Is Our Genome” (Nature 421.6921 [2003]: 409–412), Fig. 2, p. 410, https://doi.org/10.1038/nature01400 © Springer Nature Limited, all rights reserved (reproduced with permission by Springer Nature Customer Service Centre GmbH).
As a consequence, Pääbo explained as early as 2003 that “[r]ather than thinking about ‘populations’, ‘ethnicities’ or ‘races’, a more constructive way to think about human genetic variation is to consider the genome of any particular individual as a mosaic of haplotype blocks” (410). For certain haplotype blocks in the genome, an individual from Europe might be more closely related to persons in Africa or Asia than to other Europeans. Even the genetic histories of humans and other apes are “entangled” in this way, with some human genome regions exhibiting closer relations to gorillas than to chimpanzees and bonobos (Pääbo 2003, 409) (see Figure IV.24).
Thus, some researchers have argued that, regarding intra-human phylogeny, also producing a series of trees for different genome regions and then combining them into one tree of human populations is faulty, because there are no discrete entities like populations and there is no hierarchical arrangement of such geographical entities in a tree. If one takes into account all the available data to infer phylogenetic relationships, rather than a few markers considered to be particularly informative regarding ancestry, a lack of both hierarchy and discreteness is the result (and Ancestry Informative Markers were not developed for inference of population-level patterns but for the mapping of genomic regions in individuals). Molecular systematist Rob DeSalle and colleagues instead speak of “rampant polyphyly” (2017, 104), with ancestry being more about parts of the genome in different individuals that can be traced back to the same point in the history of those genomes.
Something like this seems to be at play in what was referred to as “the first ever world-wide family tree” (Currin 2022, title). Stanford statistical geneticist Anthony Wilder Wohns and colleagues (2022), including Patterson and Reich, referred to the unified genealogy of thousands of modern and ancient genomes as a tree and they started with the assumption that there was recombination. From the huge dataset of individual genomes (mainly recent and from the 1000 Genomes Project, the Human Genome Diversity Project, and the Simons Genome Diversity Project) a dated and located tree sequence of multiple correlated trees along the genome was built in an iterative approach. The researchers first merged the modern data, inferred a tree sequence for each autosome, and carried out the respective estimations. Then they integrated the archaic (Neanderthal and Denisovan genomes) and ancient samples with the modern samples and re-inferred the tree sequence. The result was seen as a step in the direction of arriving at “the genealogy of everyone” (6).
Resonating with DeSalle’s and colleagues’ “rampant polyphyly” constituted by the different parts of human genomes, Wohns et al. (2022) followed specific genome blocks back through generations, tracking how they mutated and moved (recombined, and migrated across the globe in human carriers), reconstructing the ancestral lines of the haplotype fragments found in the dataset (and inferring some twenty-seven million ancestral haplotype blocks on the way).4 While being called an “inferred tree sequence of chromosome 20” (Anthony Wilder Wohns, personal communication, 6 December 2023), Figure IV.25 suggests this new understanding of human genomic history, kinship, and diversity to be rather rhizomatic.
Have we finally grown tired of trees, as Deleuze and Guattari had? Have we arrived at levels of analytical depth in population genomics that atomize genealogy as we know it into changing rhizomatic dynamics that depend on the kind and number of data we use, and that connect parts of the genome across once closed and hierarchically related entities? Have these technologies put an end to the tree’s definitions of fixed and hierarchical subject, ‘racial’, and species positions? It is interesting that Deleuze and Guattari favored the diagram of the map, which they conceived of as an experiment on reality, as an open, undirected process. Figure IV.25 seems to have turned the tree-on-a-map image into a rhizome on a globe that is remaking the world from the Eurocentric Mercator projection to one centered around Africa, losing any sense of unidirectionality.5
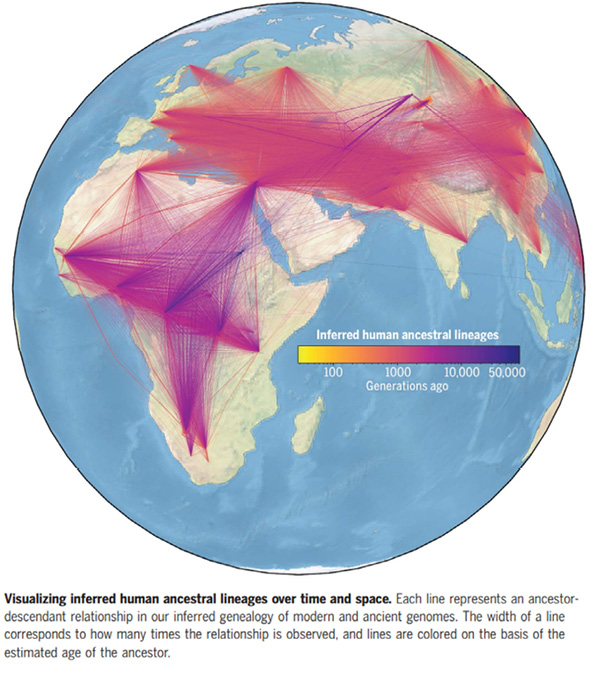
Fig. IV.25 “Visualizing inferred human ancestral lineages over time and space”. From Anthony Wilder Wohns, Yan Wong, Ben Jeffery, et al., “A Unified Genealogy of Modern and Ancient Genomes” (Science 375.6583 [2022]: 1–9), p. 1, https://doi.org/10.1126/science.abi8264. Reprinted with permission from AAAS © The American Association for the Advancement of Science, all rights reserved (“Readers may view, browse, and/or download material for temporary copying purposes only, provided these uses are for noncommercial personal purposes. Except as provided by law, this material may not be further reproduced, distributed, transmitted, modified, adapted, performed, displayed, published, or sold in whole or in part, without prior written permission from the publisher”).
Such perspectives may indeed seem liberating. To move away from the bounded notion of a species, a population, and even an individual towards understandings of greater interconnectedness may suggest a way out of identitarianism, racism, and speciesism. However, Winder and Winder (2014, 307) have cautioned that notions of a polyphyletic human species and different phylogenies for different ethnic groups might just as well feed into the kinds of racism we have observed in the preceding parts of this book. Furthermore, as we have seen, the notion of different segments of a genome having their separate genealogies can still be associated with the understanding that some segments derive from African ancestors while others derive from European ancestors, etc., which people easily link to the continental racial categories of old (e.g., Reich 2019, 147–50). Nonetheless, thinking along the lines of different parts of individual genomes having separate genealogies, connecting across what have previously been conceived as closed species, ‘races’, and organisms that have their fixed place within the hierarchical system of trees creates opportunities for new and nonhierarchical kinds of relatedness.
After all, and as mentioned at the outset of this chapter, the deconstruction of the human family tree is a special case of a larger challenge levelled at the tree of life by philosophers as well as biologists with the insight that different areas of the genome did not evolve in congruence. It was the growing knowledge of horizontal gene transfer between branches of the tree of life in nature and genetic engineering in the laboratory that served Deleuze and Guattari as an example of ‘making rhizome’: “More generally, evolutionary schemas may be forced to abandon the old model of the tree and descent”, adopting “instead a rhizome operating immediately in the heterogeneous and jumping from one already differentiated line to another” (1987 [1980], 10). Today, certainly more than ever, the assumption of a tree of life, or of a generally dichotomous phylogenetic system, is in jeopardy. We not only know that procaryotes (the vast majority of life) exhibit lateral gene transfer and that many plant and animal species hybridize. A major issue is also endosymbiosis, such as of chloroplasts, mitochondria, and other organelles of eukaryotic cells.
Therefore, while some concede that the tree retains its epistemic value as a model – and therefore hold it as unproblematic that “[c]onstructing trees is the starting point for nearly every study in evolutionary biology today” (Velasco 2012, 624; e.g., also O’Malley and Koonin 2011) as long as biologists are aware of their model-status – others advocate for a paradigm-shift towards network or web thinking also in the context of the tree of life (e.g., Doolittle 1999; Doolittle and Bapteste 2007; Bapteste and Dupré 2013; for a review of some of the network literature, see Whitfield 2012; on the controversy, see O’Malley, Martin, and Dupré 2010; O’Malley and Koonin 2011). Was “[t]he tree of life […] always a net”, because “[n]ature was always a genetic engineer” (Helmreich 2003, 351)? Such a view might well challenge traditional understandings of the organic world as falling into and being aptly represented by hierarchal relations between more or less discrete categories. Reminiscent of Darwin’s tangled bank that we have found in tension with the tree of life in Part II, such a view might rather suggest forms of relatedness that further an ecosystem-oriented thinking along the lines of coalitions and shared environmental risks (Helmreich 2003; 2009; Bapteste, Bouchard, and Burian 2012; more generally, Schmidt-Burkhardt 2009, 178). At the same time, these new kinds of biological relationships – these new kinds of relating diagrams – too, do not carry meaning in and of themselves and can be instrumentalized for different politics of life and of the human.
1 It seems that within human population genomics, however, it is the first method which might be gaining ground more easily (see below). Starting from a network and pruning it using rigorous statistical criteria is seen as a consistent inference procedure, and if a tree might correspond more closely to the information in the data, one might just as well arrive at it by this method (Templeton, personal correspondence, 8 January 2024).
2 For another example where the tree (arrived at by TreeMix) did not fit the studied population-genetic history but a network approach (program SpaceMix) led to statistically and historically satisfactory results, see, e.g., Pugach 2016.
3 See, for example, also the work of biological anthropologist Rebecca R. Ackermann, e.g., Ackermann et al. 2019. Questions regarding species concepts, species status, and the role of processes like introgression, hybridization, or continuous gene flow within the hominin line/s have a long history in paleoanthropology; for a discussion among some of the central figures, see, for example, Holliday 2003; for a more general discussion of trees and networks in connection with the program SplitsTree, see Huson and Bryant 2006.
4 What Anthony Wilder Wohns and colleagues (2022) carried out was a so-called Ancestral Recombination Graph (ARG) inference that constructs a series of trees for individual genome sites over chromosomes in a given dataset of genomes. Boundaries between trees mark ancestral recombination sites, i.e., sites at which chromosome segments that differ in their genealogies were brought together through ancestral cross-over recombination. Wohns et al. used the algorithm tsinfer. Since it assumes that the frequency of an allele is correlated with its age, it has been judged unsuitable for ARG inference involving modern human, Neanderthal, and Denisovan genomes in another study (Schaefer, Shapiro, and Green 2021, 1). This study by Nathan K. Schaefer, Beth Shapiro, and Richard E. Green from the University of California, Santa Cruz used a different algorithm (SARGE) working with shared alleles and shared inferred ancestral recombination events. They used a parsimonious approach (minimal number of necessary recombination events to allow for the data), drawing on Song and Hein (2005). A sequence of trees should be built for the sites found in the data, so that when moving along the genome, a change in tree topology would refer to a recombination. The sequence segments of a particular tree are the haplotype blocks that come with specific evolutionary histories. These trees are combined in the minimal ancestral recombination graph. Schaefer and colleagues found among other things that Neanderthal admixture could not be accounted for by a single ingression pulse into modern humans. Instead, they reached a picture of many small-scale population-specific admixture events that suggested a complex history of admixture throughout Eurasia (2021, 9).
5 Nonetheless, Wohns stated that “[k]ey findings from our paper are readily apparent [in the figure], including the ancient lineages in Africa, some ancient lineages corresponding to the archaic genomes from Siberia, generally a larger number of lineages within continents, and a few particularly ancient lineages leading to Papua from Africa and Siberia” (personal communication, 6 December 2023).