
5. Reading and organising the genome
©2025 Aswin Sai Narain Seshasayee, CC BY-NC 4.0 https://doi.org/10.11647/OBP.0446.05
5.1. Expressing the genome and decision making
The genome is a blueprint,1 and does not by itself get its host cell up and running. The genome must be read and interpreted before it can set in motion many series of connected events that somehow create life. The first step in this process is transcription, during which a gene sequence, a small part of the genome, is read and an RNA transcript with a sequence corresponding to that of the transcribed DNA is produced. Many of these RNA molecules serve as messengers (mRNA), and are further read to create proteins during translation; other RNA molecules, such as the rRNA and tRNA which help the ribosome perform translation, play direct roles in cell function without being translated. The discussion in this chapter will focus on transcription, how it is regulated, how regulators of transcription evolve and the role played by genomics in our understanding of these processes. We will conclude by asking how transcription and the manner in which genes are strung together to form a genome are linked.
Transcription is essentially an enzymatic process that is constrained by the sequence of the DNA being transcribed. The process minimally requires a DNA template, free ribonucleotides that can be linked together to form the RNA chain and an enzyme that can polymerise ribonucleotides to create an RNA sequence that is complementary to the sequence of the DNA template. In addition, the mechanics of transcription requires additional enzymes that help unwind the DNA in front of the machinery that performs transcription, and a host of other proteins that ensure that the process doesn’t stall in the middle of a gene and terminates at the right place; these will not be described much in this book. The discovery of the enzyme and that of the fact that transcription is tightly regulated in bacterial cells played important roles in the series of epiphanies that led to the explosion of molecular biology in the 1960s.2
In order to transcribe a gene, RNA Polymerase (RNAP), the enzyme that performs transcription, should specifically bind somewhere near the start of the gene. Once this happens, the double-stranded DNA must unwind and the unwound DNA must move base-by-base relative to the RNAP. As the enzyme reads the DNA bases, ribonucleotides complementary to the base being read should be assembled and attached to the growing, nascent RNA chain. The DNA in front of the RNAP must be kept unwound throughout the process. Finally, the RNAP should drop off the gene and terminate transcription at the end of the gene. The focus of this chapter will be on transcription initiation.
The RNAP is a multi-subunit protein,3 i.e., it comprises several proteins that assemble together to form a functional enzyme. These subunit proteins include those that perform the enzymatic reaction of linking ribonucleotides together, proteins that ensure that the enzyme stays on the DNA through the length of the gene and assembly factors.4 The core RNAP, which in E. coli has five subunits, is perfectly capable of performing transcription but cannot specifically recognise and initiate transcription at the start of genes. Specific recognition of these transcription start sites requires an exchangeable subunit called the σ-factor (sigma factor; Fig. 5.1). The σ-factor binds to the core RNAP, forming what is called the RNAP holoenzyme. The RNAP holoenzyme then specifically recognises DNA sequences upstream of the start of genes. The σ-factor also helps the RNAP unwind the DNA, thus initiating transcription. The σ-factor usually dissociates from the RNAP complex after initiation.
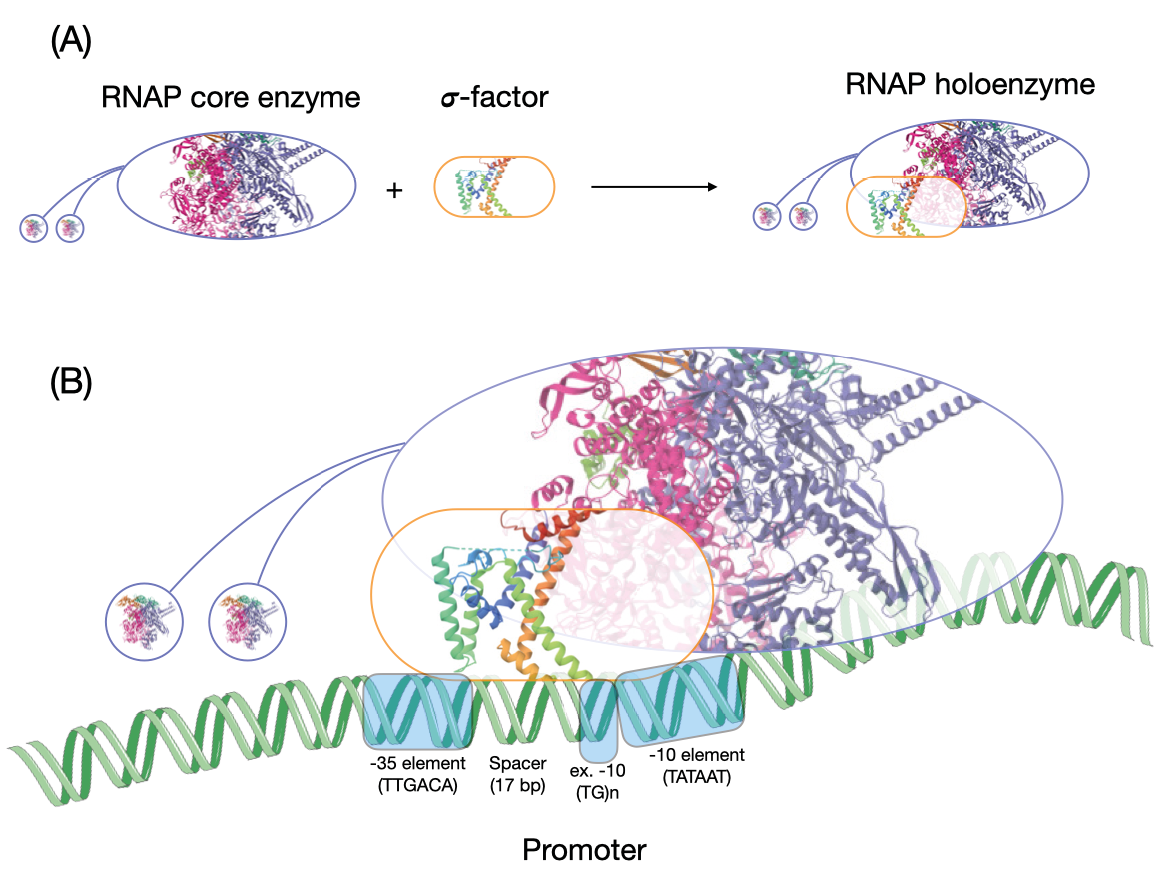
Fig. 5.1. Transcription initiation. (A) This figure shows the formation of an RNAP holoenzyme by the binding of the RNAP core enzyme with a σ-factor. The structure of the RNAP inside the oval is from PDB: 7MKP, and that of a fragment of a σ-factor is from PDB: 1SIG. (B) This figure shows the interaction of an RNAP holoenzyme with the promoter. The image of the DNA is from SMART-Servier Medical Art, part of Laboratoires Servier, via Wikimedia Commons, available freely under CC BY-SA 3.0.
The DNA sequence that the RNAP holoenzyme recognises is called the promoter. The promoter region is usually A+T-rich. Each gene has its own promoter sequence, but taken together many promoters show some common properties. For example, the bacterial promoter—based on the paradigm established in E. coli but shown to be applicable to many other bacterial genomes5—is bipartite. There is a six-base −10 element (minus 10) and a six-base −35 element (minus 35). The −10 element is centred 10 bases upstream of the transcription start site (the site at which mRNA synthesis begins) of a gene, and the −35 element is centred 35 bases upstream. The −10 element, when analysed across many genes, has a consensus sequence TATAAT, whereas the −35 element shows a consensus of TTGACA. The specificity-determining σ-factor, when bound to the RNAP, recognises these elements on the DNA. The sequence of the stretch of DNA between the two elements is immaterial. However the length of this spacer is critical to ensure that the −10 and the −35 elements are oriented correctly for the RNAP to bind to the promoter. The precise consensus sequence is not necessary to produce a functional promoter. It is merely a construct that represents the most common base found at each site.
Natural promoters usually differ from the consensus at one or more sites, and the more divergent it is from the consensus element the weaker is its affinity to the RNAP. Therefore, each gene, on the basis of its promoter sequence alone, has its own unique ability to attract RNAP and initiate its own transcription. This creates cross-gene variation in the extent to which a gene can be transcribed. Some promoters do not contain a −35 element, and these sequences carry what is an extended −10 element, which is a slightly longer version of the −10 sequence motif.
Though the sequence of the promoter itself can determine to some extent the expression level of a gene, this does not vary within the lifetime of a cell. Changes in the promoter sequence can happen over generations and, similar to mutations within a gene sequence, its fate can be determined by selection or drift. However, a cell often needs to make decisions in a matter of minutes about which gene to express, and when, within its lifetime. Many bacterial cells experience conditions that change from time to time. Even if their genetic repertoire is sufficient to handle all these environmental conditions, only a subset of their genes would be required under any given condition. Expressing the rest can be costly. As we noted in Chapter 3, expressing a gene under conditions in which the gene offers no selective advantage to the cell can be very costly, especially in bacteria with large population sizes. In addition, there is a constraint that arises from resource availability. The number of free RNAP molecules available to initiate transcription is often limited, because ~80% of all RNAP molecules are involved in transcribing a very small number of genes coding for rRNAs.6 Therefore, the number of RNAP molecules available for transcription is often far less than the number of genes in the genome. Finally, in complex bacterial genomes, the expression of one gene may counteract that of another and such conflicts should necessarily be contained. Thus, regulation of gene expression, or, in other words, taking decisions on which gene to express at any point in time, is important.
Various regulatory systems, or networks, help the cell achieve gene regulation. First, though the sequence of DNA is relatively static, its structure is not. The DNA double helix is usually in what is called a B-form, in which each turn of the DNA has ~10 base pairs. The double helix can unwind or overwind such that the number of base pairs per turn is less than or greater than 10, and, in E. coli, this is to a large extent determined by the energy levels available to the cell.7 DNA that is unwound is said to be negatively supercoiled. As the degree of negative supercoiling decreases and approaches the standard B-form twist, the DNA is said to be more relaxed. E. coli DNA is rarely, if ever, positively supercoiled, though this is known to happen in other bacteria. Enzymes under the umbrella name topoisomerase help modulate supercoiling states of DNA. In E. coli a topoisomerase called DNA gyrase negatively supercoils DNA, whereas DNA topoisomerase 1 helps relax DNA. When the cellular energy levels are high, the DNA is negatively supercoiled due to high DNA gyrase activity and this permits rapid transcription; during starvation, the DNA becomes relaxed, which can globally suppress transcription.8 However, this overarching link between DNA supercoiling and transcription does not apply equally to all genes (Fig. 5.2). It has been observed that genes whose expression is preferentially reduced during starvation (or whose expression is high specifically during rapid growth) have a G+C-rich region in their promoters. This might make the promoter harder to unwind because G-C base pairs are more stable than A-T base pairs.9 Unwinding of such promoters might be facilitated by negative supercoiling, which is favoured during high growth states. This mechanism appears to affect the expression of many genes involved in translation, including that of rRNA, whose transcription at high levels under nutrient stress can be hugely wasteful and damaging. Under nutrient-replete conditions, however, high transcription of such genes is necessary to support growth. On the other hand, promoters that are extraordinarily A+T-rich may be preferentially transcribed during starvation, when the genome in general is less negatively supercoiled.10 Therefore, the fact that the structure of the DNA can respond to some cellular conditions and in turn affect the extent to which different genes are transcribed makes the DNA itself an important regulator of gene expression.
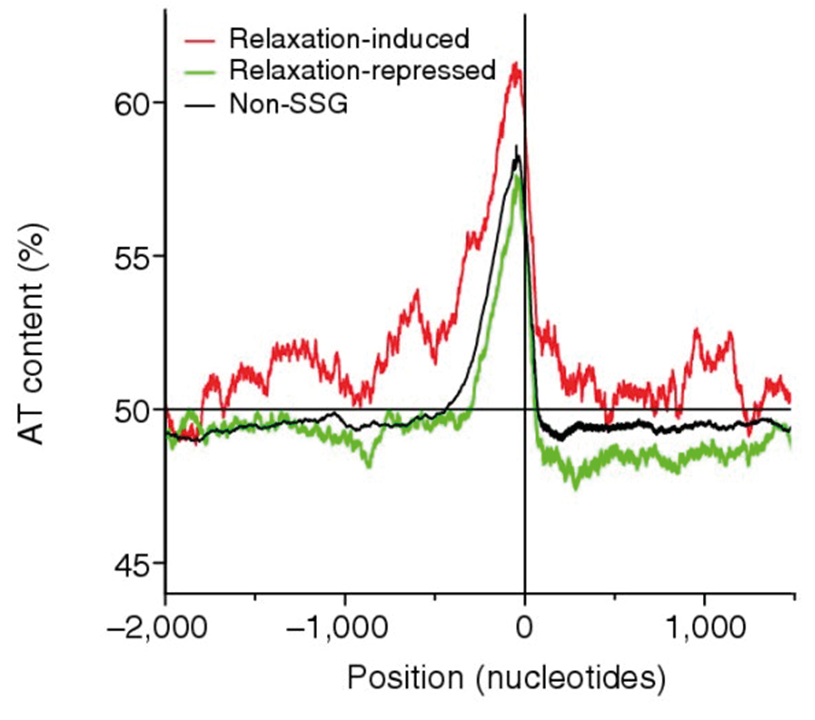
Fig. 5.2. Base composition upstream of genes regulated by DNA supercoiling in E. coli. This figure shows that genes that are induced by DNA relaxation are more A+T-rich than the average gene, whereas the reverse holds for genes that are induced by negative supercoiling. Along the x-axis, values to the left of ‘0’ indicate positions upstream of genes, and positions to the right indicate the gene body and further beyond. Originally published as Figure 5B in B.J. Peter, J. Arsuaga, A.M. Brier, A. Khodursky, P.O. Brown, and N. Cozzarelli, ‘Genomic transcriptional response to loss of chromosomal supercoiling in Escherichia coli’, Genome Biology 5 (2004), R87, CC BY 2.0.
Yet another component of the core transcriptional machinery that plays a regulatory role is the σ-factor, which helps the RNAP recognise promoters. Many bacterial genomes code for multiple σ-factors. Depending on the relative abundance of the core RNAP and σ-factors, the multitude of σ-factors can all be bound to abundant RNAP molecules, or they compete for the limited real estate presented by an insufficient number of core RNAP molecules. Though protein quantification by different labs support different scenarios, the most comprehensive and recent analysis (to my knowledge) supports the latter.11 Thus, we now accept that different σ-factors compete with each other for binding to the core RNAP. The outcome of this competition will be determined by the relative abundance or availability and the affinity of each σ-factor to the core RNAP.
Different σ-factors recognise different promoter types. The standard bipartite promoter structure that we described earlier is best recognised by what is called the σD σ-factor, following the nomenclature used for E. coli. σD is a ‘housekeeping’ σ-factor that, by recognising the standard promoter, helps initiate transcription of a majority of genes involved in growth and metabolism that operate in nutrient-rich conditions. A second σ-factor, σS in E. coli, becomes available in sufficient concentrations as nutrients deplete and cells enter a period of starvation and stress. This σ-factor helps the RNAP bind to promoters of genes underlying the bacterial response to a variety of stresses, which together form the general stress response. There is evidence that several promoters bound by σS-bound RNAP are recognised by this σ-factor when the DNA is relaxed, as it is during starvation, pointing to how DNA structure can contribute to differential promoter recognition by various σ-factors.12 Because different σ-factors, when bound to the RNAP, can recognise their own set of promoters, the outcome of the competition between these σ-factors for binding to the core RNAP is a major determinant of which genes are expressed. We will return to this aspect of regulation later in this chapter.
Whereas the structure of the DNA and σ-factors (as regulatory molecules) are still part of the machinery that performs transcription, several other ‘outside’ players fulfil important roles in gene regulation.13 The most prominent among these are transcription factors (TFs). TFs are DNA-binding proteins. They bind to specific DNA sequence motifs often present around the promoter region. The DNA sites to which TFs bind are sometimes called operators, or simply TF-binding sites. When bound to these sites, TFs can either activate or repress transcription (Fig. 5.3).14 TFs repress transcription usually by binding close enough to the promoter that they block access to the RNAP; in other words, they sterically hinder RNAP-promoter interactions. By binding to one site near the promoter and another further upstream, they can also loop the intervening DNA and form a strongly repressive structure that prevents RNAP activity. Sometimes they do not block the initial interaction between the enzyme and the DNA, but instead prevent further progress.
The discovery by Arthur Pardee, Francois Jacob and Jacques Monod of a repressor of the set of genes that help E. coli metabolise sugar lactose, published in 1959, played a central role in the discovery of messenger RNA.15 This repressor was isolated a few years later by Benno Muller-Hill16 and shown to bind specifically to its operator site on DNA. While Monod was working on the induction of lactose metabolism genes, Andre Lwoff was demonstrating the phenomenon of bacteriophage lysogeny. Mark Ptashne’s work revealing the central role of repressors of transcription in the maintenance of lysogeny was yet another landmark in the history of gene regulation.17
The discovery of the repressor-based regulation of lactose metabolism genes also showed that in bacteria several genes encoded in tandem on the genome can be expressed from a single promoter. Such a series of co-transcribed genes is referred to as an operon, a fundamental feature of bacterial genomes: the ~4,000 genes in the E. coli genome may be organised into ~2,000 operons. Not all genes are organised into operons; many are singletons. Some operons are short, comprising not more than two or three genes whereas other uber-operons can encompass tens of genes.
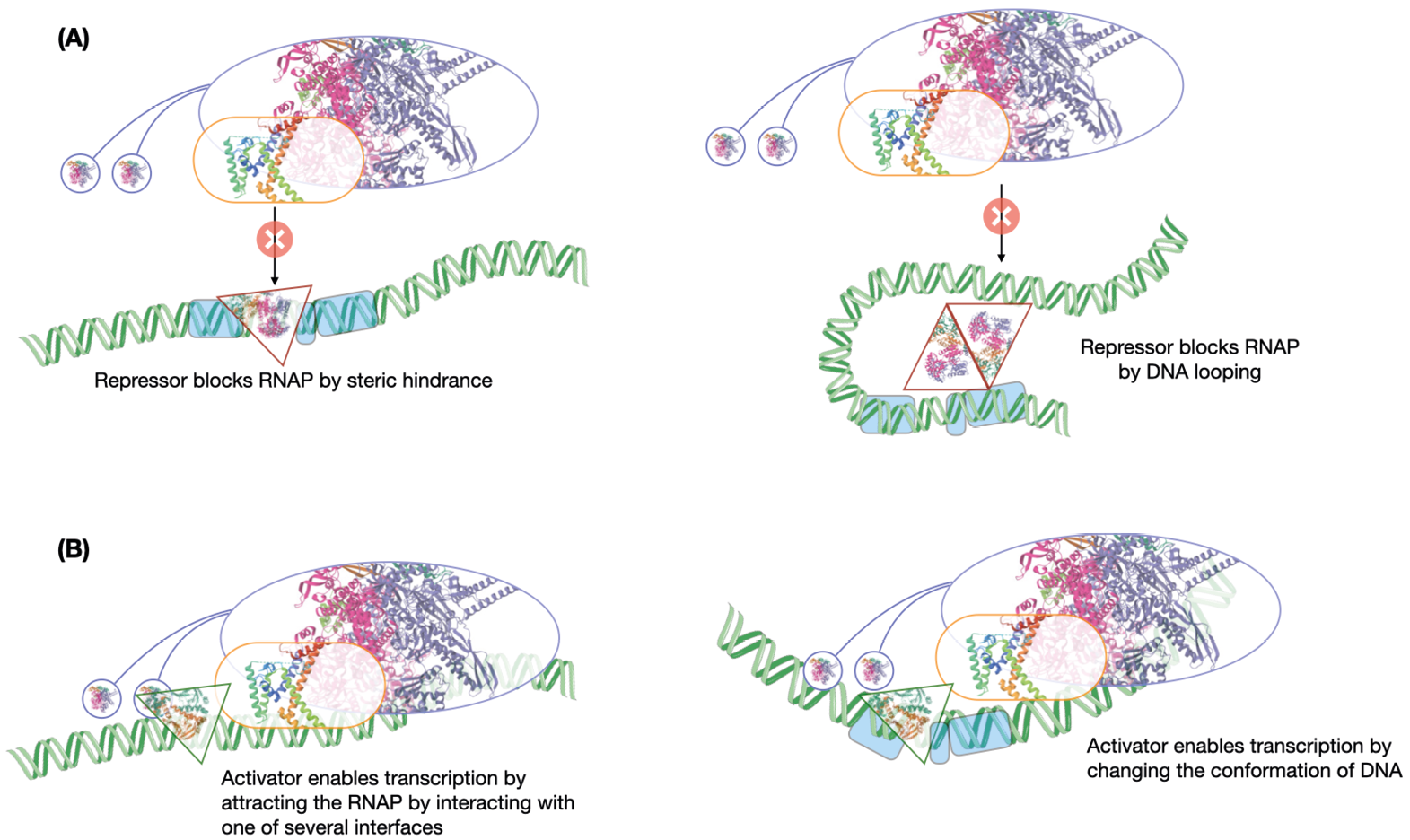
Fig. 5.3. Activation and repression by transcription factors. This figure shows a sample of simple ways by which (A) repressors (red-bordered triangles) and (B) activators (green-bordered triangles) act to regulate transcription initiation. The structure of the lac repressor filling the red triangle is from PDB: 1LB1, and that of CRP filling the green triangle is from PDB: 4N9H. The images of the DNA are from SMART-Servier Medical Art, part of Laboratoires Servier, via Wikimedia Commons, available freely under CC BY-SA 3.0.
Not all regulators of transcription are repressors. Activators normally bind upstream of the promoter and have elements that can attract the RNAP, interacting either with the core RNAP components or with the σ-factor. Ellis Engelsberg, along with his colleagues Joseph Irr, Joseph Power and Nancy Lee, discovered in 1965 that the genes for utilisation of the sugar arabinose in E. coli came under the control of an activator.18 This discovery was initially met with much scepticism because of the deeply entrenched repressor-based model of gene regulation espoused by work of Pardee, Jacob and Monod. In the words of Steven Hahn, “(though) the evidence in 1965 for positive control by AraC was as good or better than the data used to formulate the negative control model, Englesberg needed to accumulate much additional data to answer his critics.”19 Nevertheless, it was only a matter of time before other activator systems were described and Engelsberg stood vindicated. Many TFs can activate transcription of one gene but repress that of another, and whether they activate or repress transcription depends on where they bind relative to the promoter.20 Some TFs, including Engelsberg’s activator, can even perform dual actions on the same target gene by changing its binding site upstream of the gene. One can expect any activator to be able to repress transcription as long as it binds the DNA in such a manner that the RNAP cannot bind to the promoter. The reverse need not be true—a pure repressor cannot activate transcription just because it binds upstream of the promoter—it may not possess an interface to attract the RNAP.
The activity of TFs themselves is often determined by the presence or absence of a signal. For example, a TF that activates transcription of genes responsible for metabolising a sugar as a nutrient will be activated by the presence of the sugar. Such a TF, in addition to being able to bind DNA, will also be able to bind to the sugar to which it responds. The binding of the sugar to the protein will then activate (if the TF is an activator of the sugar metabolism genes) or hinder the TF’s ability to bind to the DNA (if the activator is a repressor). Many TFs in bacteria possess such a property. The repressor of lactose metabolism binds to allolactose (similar to lactose). When not bound to allolactose, the repressor binds to the DNA and blocks RNAP activity. The binding of allolactose causes the TF to release the DNA, thus allowing transcription. The activator of arabinose metabolism binds to the DNA both in the presence and absence of the sugar. In the former situation, it acts as an activator but switches to being a repressor in the latter. Other TFs may not directly bind to a signal, but may be activated following a series of reactions that are initiated by a separate signal-sensing protein that, for example, may be located on the cell membrane. Each TF regulates its own set of target genes and the set of TF-target gene interactions constitutes a transcriptional regulatory network. Thus, TFs are proteins whose activities are usually determined by the presence of certain environmental or cellular conditions, in response to which they regulate the transcription of other genes.
5.2. The transcriptional regulatory network of E. coli
The E. coli genome encodes ~300 TFs for its total complement of ~4,400 genes. Even in this well-studied organism, we do not know all the regulatory connections these TFs make. Over half of these TFs have at least one known target gene,21 as discovered through biochemical or genetic experiments; the others are predicted to be TFs based on their sequences. In addition to these TFs, E. coli has seven σ-factors competing to bind to the core RNAP. These TFs and their target genes or operons together constitute the transcriptional regulatory network. For E. coli there are publicly available databases such as RegulonDB22 and Ecocyc,23 from which the currently known transcriptional regulatory network can be downloaded. These networks comprise of data from a variety of experiments—from small-scale, detailed studies on how a particular TF binds to an operator to regulate a target gene, to large-scale, bird’s-eye view studies that catalogue the list of all genes or operons that are regulated by one or more TFs under a set of growth conditions.
The targets of a TF can be defined in several ways. A gene can be called a target of a TF if the regulator binds upstream of the gene and, when bound, alters the expression state of the gene. This would define direct targets of a TF. Sometimes, the mere binding of a TF to an operator is used to define a target irrespective of whether there is evidence that the binding affects the expression of the gene. This may be appropriate when there is reason to believe that absence of evidence (of an effect on gene expression) is not evidence of absence. On the flip side, some TF-DNA interactions may also be non-functional. Alternatively, genes that change in expression when a TF is deleted can be called targets of the regulator. However, the targets defined may, therefore, not always be bound by the TF, and may change in expression as a result of a cascade of effects initiated far upstream by the direct regulation of a different gene(s) by the TF.
Given such complications in the ways in which a regulatory network can be defined, are such networks even useful to define on a genome-wide scale? In other words, does a regulatory network—built by aggregating data from hundreds to thousands of experiments together encompassing a cocktail of approaches—predict gene expression: the defining, measurable output of the regulatory network? Xin Fang and colleagues recently showed that a transcriptional regulatory network built from data on where TFs bind on the genome agrees well with genes that change in expression when a TF is deleted, and that the regulatory network is good enough to predict the gene expression states of over 85% of operons.24 Earlier work by Gabor Balazsi and colleagues had shown that groups of genes that are expressed together under a given condition often belonged to coherent, closely-linked parts of the then known regulatory network.25 Thus, the transcriptional regulatory network—despite being incomplete even for a well-studied model organism such as E. coli—serves as a good predictor of gene expression. However, though groups of genes regulated in the same manner may be expressed together, the expression level of a TF may not correlate well with that of its targets, in part because the activity of a TF is not defined entirely by its expression level.26
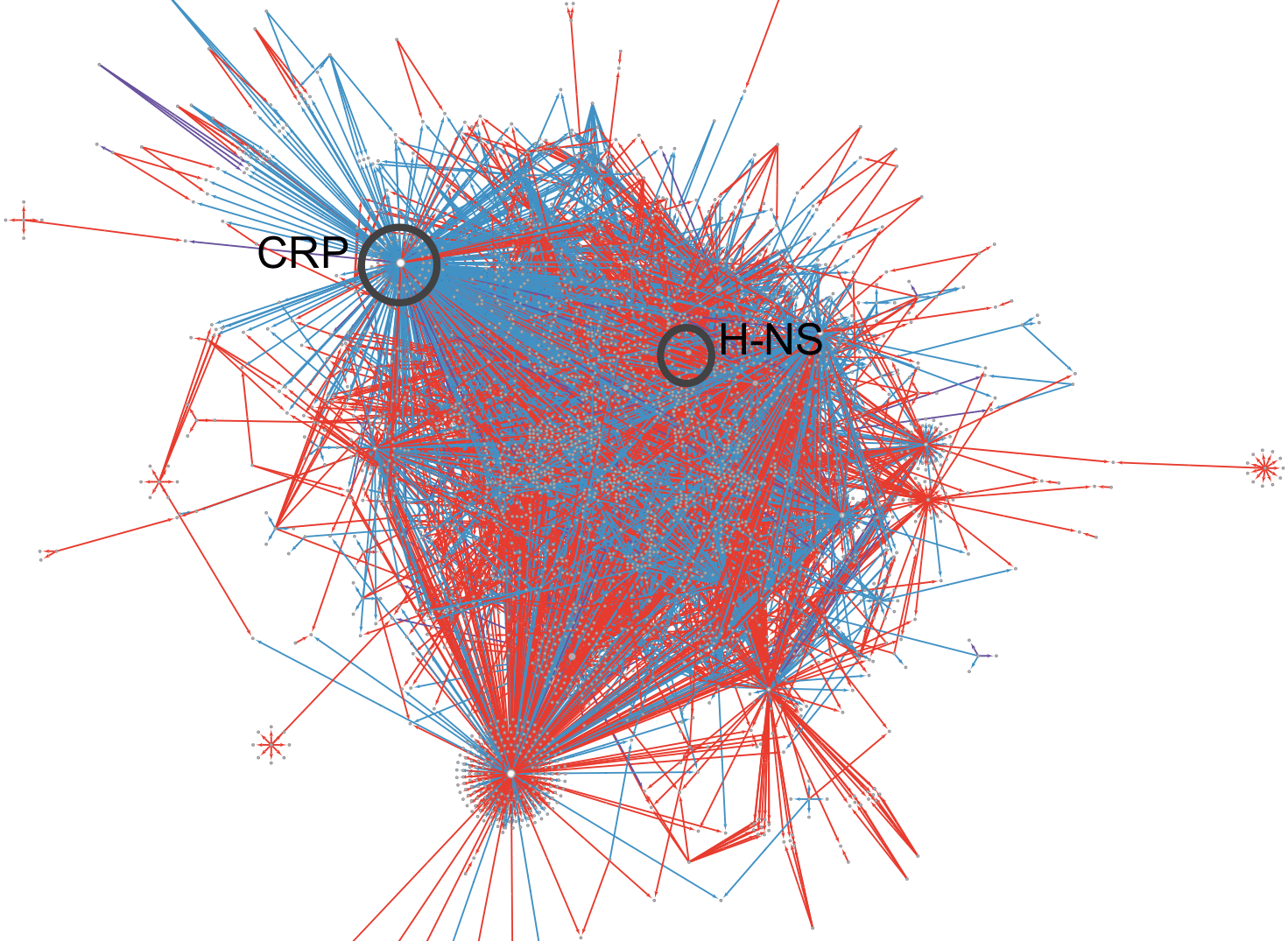
Fig. 5.4. The E. coli regulatory network. This figure shows a representation of the E. coli transcriptional regulatory network. Each line indicates a regulatory interaction between a regulator (mostly, but not necessarily, TFs) and a target gene. Red lines mark repressive interactions, whereas blue lines indicate activating interactions. Global regulators CRP and FNR are marked. Figure produced by Ganesh Muthu using the regulatory network available in the RegulonDB database (https://regulondb.ccg.unam.mx/) and Cytoscape (y-Force layout; https://cytoscape.org/ and https://www.yworks.com/products/yfiles-layout-algorithms-for-cytoscape).
The first decade of this century saw the publication of several papers describing graph theoretical studies of biological networks. Among these networks are transcriptional regulatory networks. To some extent, these studies were spurred by genome-scale studies of the eukaryote Saccharomyces cerevisiae, a yeast. One major work identified binding sites for over 150 TFs encoded by this organism,27 triggering a large number of studies curating and analysing the vast amounts of data produced by this work. Any network is a graph that draws edges connecting points called nodes. In what is called a protein-protein interaction network, nodes are proteins and an edge is drawn between two proteins that physically interact with each other. The edges in such a network are not directional: if protein A interacts with protein B, then B also interacts with A and there is no direction to how the two proteins interact with each other. A transcriptional regulatory network, which connects TFs to their target genes or their binding sites, is directional: each edge is directed from the TF to a target gene because the TF regulates the expression of the target gene (Fig. 5.4). Some representations of the transcriptional regulatory network also include σ-factors as regulatory proteins similar to TFs, others do not.
As one would immediately guess, such networks are not exclusive to biology. One can envisage a whole host of networks—such as the internet, electricity grids, and postal networks—and all of these can be directional. In, say, a postal network, which connects two post offices with an edge if letter bundles are sent from one to the other, not all nodes are connected equally. For example, the general or hub post office of a city will receive all letters sent to the city and then forward it to various local offices. The local post offices, despite being in the same city, may not be connected to each other directly. Thus, many post offices will have low connectivity, often being connected both ways with only the city’s general post office. The general post office on the other hand is highly connected, but the number of such offices is very small compared to the number of local offices.
Similar trends have been described for biological networks as well. For example, in the E. coli transcriptional regulatory network, most TFs regulate only a few genes;28 the repressor of lactose metabolism regulates only what is called the lac operon. On the other hand, a few TFs regulate hundreds of genes! The former, with their limited sphere of influence, are often called local TFs, and the latter, in contrast, are referred to as global TFs. That said, however, the distribution of the number of targets a TF has is continuous, and therefore it is not entirely obvious where a line demarcating local from global TFs should be drawn. As a result, there have been several definitions of what constitutes a global TF.
Often, an arbitrary threshold number of targets is used to separate global and local TFs. But is the number of targets the only parameter that defines global TFs? Some studies, primarily by Julio Collado-Vides and colleagues, argue otherwise. To follow this line of thinking, we must first understand and visualise the network itself a bit better. The regulatory network is not a disconnected set of TFs regulating their target genes in a one-on-one or a one-on-many manner. Just as a TF can regulate multiple genes, many genes are regulated by multiple TFs. For example, the operon for lactose metabolism is regulated not only by the lactose-responsive repressor but also by a TF CRP that responds indirectly to glucose availability in such a way that CRP becomes active when glucose is limiting. The same CRP acts as a second regulator of arabinose metabolism as well. The lactose operon is expressed only when the repressor is not bound and CRP is bound; the arabinose metabolism genes are expressed when the arabinose-responsive TF is bound to its operator sites in an activating configuration and CRP is also bound. Genes that determine the decision of the E. coli cell to move or to stay put are regulated by several TFs, and biochemical experiments with purified protein and DNA sequences suggest that several tens of regulators can bind to regions upstream of these genes.29 It is well-nigh impossible for all these regulators to bind simultaneously to the small stretch of DNA upstream of these genes determining motility/adhesion. However, different small sets of regulators may be active and involved in the regulation of these genes under different conditions.
Next, TFs can regulate genes for other TFs. This can set up a variety of network motifs. It can create cascades in which a series of regulatory events ultimately determines the expression of a non-TF target gene. For example, a dimeric TF called FlhDC activates the expression of a σ-factor called FliA. FliA then regulates the expression of a host of genes that allow the bacterial cell to move in particular ways. But then, the cascade is not purely linear along a single path. FlhDC, in addition to regulating the gene encoding the σ-factor FliA, also directly activates genes that allow the cell to move. This creates what is called a feed-forward loop: TF A regulates the expression of TF B, and A and B together regulate the expression of a non-transcription-factor target gene C. Depending on whether the effect of A on C is the same as the composite effect of A and B on C, the feed-forward loop is either coherent or incoherent.
Most feed-forward loops in the E. coli regulatory network appear to be coherent.30 FliA, being a σ-factor, activates all its targets and FlhDC activates its target genes. Therefore, both routes to the motility-determining genes are activating, so this represents a coherent feed-forward loop. The two activating regulatory arms leading to C may represent an AND gate, in which both arms are required for the full expression of C, or they may form a SUM gate in which the effect on C is the sum of the effects of the two individual arms. The regulatory system for motility forms a SUM gate.31 The arabinose system also includes an AND feed-forward loop in which CRP sits as a top-level TF that regulates the expression of the arabinose-responsive TF as well as that of the enzymes that metabolise arabinose. OR gates between the two arms of a feed-forward loop are also possible. For example, a coherent feed-forward loop forming an OR gate regulates the expression of a negative regulator of an adhesive structure called holdfast in a bacterium Caulobacter crescentus.32 Whereas the activating SUM input of a coherent feed-forward loop keeps the expression of motility genes on for a long time, a coherent OR input structure helps to decrease the effect of environmental fluctuations on the expression of the ultimate target gene. Other types of local network structures include one in which the same TF regulates multiple genes, but with varying binding affinities such that some targets are prioritised for regulation over others.33
Finally, feedback loops also occur. If one were to include the signal molecule, such as lactose (or allolactose), in our representation of the regulatory network, the regulation of the lactose operon would be an example of a positive feedback loop. In this positive feedback loop, activation of the lactose operon in a cell by a small quantity of the signal would result in more of the signal molecule being taken up by this cell. When lactose is limiting such that not all cells see lactose first, a bistable state, in which two sub-populations of cells with very distinct levels of expression of the lac operon will be established. The lucky few cells that came into contact with lactose early would be primed to take up most of the lactose available in the local environment, whereas others would be oblivious to the availability of this sugar. Here we immediately notice a situation where genetically identical cells display two distinct traits because of how their regulatory network has interpreted the environment. This highlights the important argument that the genome sequence is not a dictatorial directive but, as the science writer Philip Ball puts it, it helps to establish some “flexible rules” from which life can emerge.34 Negative feedback loops also exist, and there are also instances where one TF regulates another and the latter returns the favour. In other words, even if we do not include the signal molecule in our representation of the regulatory network, feedback loops involving TFs and no other types of molecules exist.35 Finally, TFs can auto-regulate their own expression, often negatively, but also positively. Whereas the former helps to reduce response time and decrease fluctuations, the latter does the opposite. Thus, each type of network motif has its own kinetic properties.36 Taken together, the take home message from this short discussion is that the transcriptional regulatory network is a complex set of highly interconnected nodes that form a variety of converging, diverging, and even circular loops.
Back to global regulators: do some regulators with a large number of targets possess additional properties that distinguish them from local TFs that regulate a small set of genes on demand? Agustino Martinez-Antonio and Julio Collado-Vides found that some TFs with a large number of targets share certain properties which together qualify them as global regulators. Firstly, they regulate genes belonging to distinct functions.37 For example, CRP regulates genes involved in carbohydrate metabolism as well as the genes encoding FlhDC, the master regulator of motility, among others. In contrast, TFs with only a few targets often regulate genes which all or mostly belonging to a single pathway or function. For instance, the TF TyrR regulates a small number of operons, all encoding genes involved in the metabolism of aromatic amino acids. In a more recent work, Julio Freyre-Gonzalez and colleagues suggested that global regulators integrate multiple modules within the transcriptional regulatory network.38 Modules are groups of highly interconnected nodes, such that nodes within a module are more connected to each other than nodes from across modules. Modules are often determined by one or more TFs with local scope whereas global regulators sit astride multiple modules, presumably priming the expression of a large and diverse set of genes. Each module would, in some ways, represent a functionally coherent set of genes, and therefore Freyre-Gonzalez and colleagues’ work might be taken as an independent validation of Martinez-Antonio’s suggestion that global TFs regulate genes from multiple functions.
The E. coli genome encodes seven σ-factors. Each σ-factor helps to transcribe its own set of genes. Though there is some overlap between the sets of genes regulated by different σ-factors, one can say that the transcriptional space of E. coli, or for that matter that of many bacteria with large genomes, is partitioned among σ-factors.39 Global TFs often regulate genes from different σ-factor partitions. According to the data analysed by Martinez-Antonio and co-workers, CRP regulates genes from as many as four σ-factor partitions! Global TFs do not usually regulate a gene as its sole regulator; they often act in concert with other TFs. Again, the regulation by CRP of its targets in sugar metabolism in concert with local TFs is a good example. Global TFs often regulate other TFs, something that TFs with a local scope rarely do. Finally, global TFs are also active in multiple conditions, whereas local regulators are usually activated by highly specific signals. Based on these parameters, Martinez-Antonio and Collado-Vides concluded that the E. coli genome encodes seven global TFs, and that a majority of target genes have at least one of these seven TFs as their regulators.
Years ago, I was involved in a piece of work that attempted to study how genes involved in metabolism are regulated in the transcriptional regulatory network of E. coli, and how different segments of the metabolic network might be regulated differently by global and local TFs.40 The metabolic network can be visualised as an hourglass. A great diversity of nutrient breakdown pathways converge down to what is called central metabolism, which eventually produces energy. And a variety of biosynthetic pathways diverge away from these central metabolic pathways. Usually, nutrient breakdown pathways are regulated by TFs that respond to the nutrient itself, i.e., these are regulated by supply levels. On the other hand, biosynthetic pathways are regulated by TFs that bind to final product molecules, i.e., these are controlled by demand. These TFs are often local. Central metabolic pathways, in contrast, are regulated by a multitude of global TFs. This makes sense in light of Martinez-Antonio and Collado-Vides’ work showing that global TFs respond to a wide range of environmental and cellular conditions, as well as the notion that central metabolic pathways are the culmination of a whole range of metabolic cues.41 Whereas biosynthetic pathways are usually regulated by a single TF, the regulation of breakdown pathways often involves combinations of global and local TFs, as exemplified by CRP acting as a major node regulating a whole variety of breakdown pathways in concert with local TFs responding to particular nutrient molecules.
Finally, global TFs are usually expressed at high levels in the cell, and this presumably is to ensure their availability in sufficient concentrations to bind to all their targets. In contrast, local TFs are expressed at low levels; in fact, there is a positive correlation between the expression level of a TF and the number of targets it regulates. As shown by Grigory Kolesov and colleagues, for a TF present at low concentrations, finding one or a few target binding sites in the cell can be inefficient.42 To counter this, local TFs are often encoded adjacently to their target sites. This logic works for bacteria in which transcription and translation occur more or less simultaneously, but not for eukaryotic cells in which transcription occurs within the nucleus whilst proteins are synthesised outside and so the TF will have to be transported back into the nucleus before it can bind to and regulate its target. There is also an evolutionary explanation for why local TFs are encoded close to their cognate targets. Horizontal transfer of a stretch of DNA carrying genes for a metabolic pathway is more likely to be successful if it also carries its own TF!
In summary, the transcriptional regulatory network—as exemplified in E. coli—is a complex structure with highly interconnected regulators and their targets. This structure ultimately determines which genes are expressed in the cell and when. Many regulators act in an intuitive manner, responding to a signal and regulating genes that should respond precisely to the inducing signal. In contrast, there are global TFs that integrate multiple segments of the metabolic network, and whose significance is a lot harder to understand and rationalise. In the next few sections, we will describe and try to understand the functioning of two regulatory networks determined by global TFs.
5.3. Driving the stress response: σS and its competition with σD
In E. coli, the major σ-factor σD regulates the expression of most genes involved in growth. It is the most abundant σ-factor protein in the cell and, out of the seven σ-factors encoded by the E. coli genome, it has the highest affinity for binding to the core RNAP. Its function contrasts with that of σS, the stress responsive σ-factor, whose production increases as nutrients deplete and growth starts to cease. σS is also expressed during growth, but under conditions that are not ideal for growth of the bacterium. These include conditions of suboptimal osmolarity, temperature, and pH.43 Like the global regulator CRP, σS does not respond nor activate cognate responses to specific stresses but plays a “preventive”44 role, priming the cell to tolerate a wide range of stresses. Given its ability to contribute positively to cellular responses to such stresses, it is not surprising that the gene for σS was discovered independently multiple times by several researchers in the 1980s–early 1990s45 before it was recognised that all these researchers had been referring to the same protein doing its job in different contexts!46
The expression of σS is regulated by a plethora of mechanisms at every conceivable step in gene expression, from transcription through translation to protein stability.47 Transcription of rpoS, the gene encoding σS, increases during down-shifts in growth, most notably as an E. coli culture transitions from exponential growth to a stationary phase.48 There is evidence that the global TF CRP is involved in the up-regulation of σS during the stationary phase. The RegulonDB database for transcriptional regulatory interactions in E. coli also includes an acid stress regulator called GadX as a regulator of the σS gene. The small molecule guanosine tetraphosphate, which is produced during transition to states of starvation, also up-regulates the transcription of the σS gene. The concentration of the mRNA for σS also decreases in the absence of the cytosine methyltransferase Dcm (see Chapter 4). Various other regulators of σS expression have been described in other bacteria as well. However, the amount of σS mRNA is not a good predictor of the amount of σS protein: σS protein has been reported to be hardly detectable under some conditions in which the σS mRNA is abundant.49 This suggests regulation of this gene beyond transcription. Some RNA binding proteins such as Hfq play roles in enabling translation of the σS mRNA in concert with other proteins and RNA that act as specific stress signals.50 Even the protein HU, best known as a non-specific DNA binding protein, may bind to the σS mRNA and affect its translation.51 The stability of σS protein is also regulated by complex signal cascades involving multiple proteins. Protein stability is a pivot point in σS expression that is targeted by adaptive strategies that use genetic evolution of σS, which we will return to later in this chapter. Thus, regulation at multiple stages of gene expression and protein stability seems to play a role in determining σS levels under different conditions. Whereas regulation at the level of transcription seems to predominate during steady transition to slow growth rates, stresses such as high osmolarity and low temperatures seem to particularly stimulate σS translation.
Harald Weber and colleagues identified genes regulated by σS in E. coli growing in three different conditions (stationary phase, osmotic, and acid stress) by measuring changes in the levels of the mRNA of all genes encoded by the genome in the presence and absence of an intact rpoS gene.52 Of the ~500 genes that changed in expression in a σS-dependent manner in at least one of the three conditions, a third were defined as the ‘core’ σS regulon, being responsive to σS in all three conditions. The members of the core σS regulon typically contained an extended −10 element in their promoters, whereas the rest did not and might be expressed from sub-optimal promoter elements by σS-containing RNAP holoenzyme, in concert with other stress-responsive regulatory proteins. Byung-Kwan Cho and colleagues, while assembling a network of regulatory interactions between all σ-factors and their targets in E. coli (Fig. 5.5), showed that σS was bound to over 1,000 promoters in E. coli, but a majority of these interactions did not result in a change in gene expression when σS was removed.53 The role of these binding interactions, if any, is yet to be understood. Whereas most of the genes whose expression changes in response to σS are activated by the σ-factor, the rest are in fact less expressed when σS is present. How could this be possible? Cho et al. showed that in many of these genes, the presence of σS reduced the binding of σD, the major house-keeping σ-factor. This suggests that competition between σ-factors, through which the presence of one σ-factor affects the influence of another, plays a role in determining the mRNA levels of several genes.
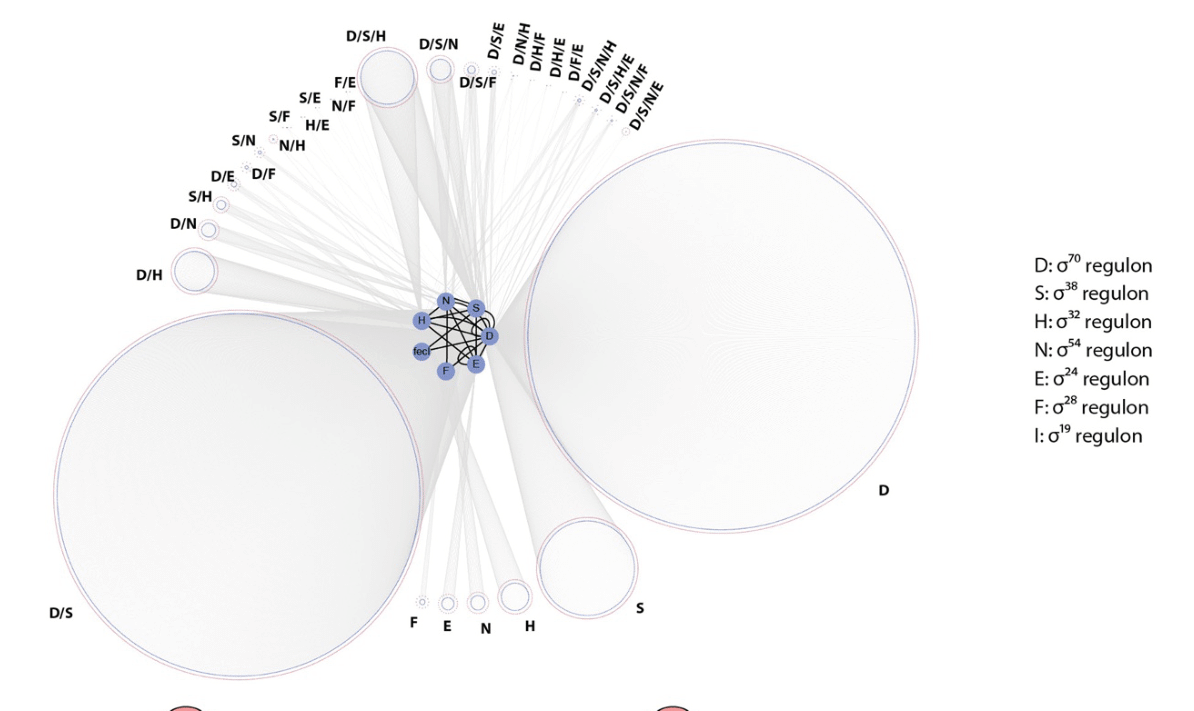
Fig. 5.5. Targets of various σ-factors in E. coli. This figure shows the sizes of various regulons (sets of targets of a regulatory protein) for E. coli σ-factors. σ38 is an alternative name for σS, and σ70 for σD. Originally published as Figure 2D in B.-K. Cho, D. Kim, E.M. Knight, K. Zengler, and B.O. Palsson, ‘Genome-scale reconstruction of the sigma factor network in Escherichia coli: topology and functional states’, BMC Biology 12 (2014), 4, CC BY 2.0.
For a σ-factor to activate the transcription of its targets, it first has to outcompete with other σ-factors for binding to the core RNAP; only then do holoenzymes bind to their respective promoters. Competition between σ-factors for a limited core RNAP—particularly the RNAP between σD and σS, both of which regulate large numbers of genes—can present some interesting problems. Though the concentration of σS increases as E. coli cells transition from exponential growth to a stationary phase, the absolute concentration of σS stays well below that of σD. In addition, the affinity of σS to the core RNAP is much less than that of σD. Therefore, even in stationary phase, σS, on its own, would be hard-pressed to compete effectively with σD to form the RNAP holoenzyme. According to calculations using the known total concentrations of σS, σD, and the RNAP in free and complexed forms, as well as the affinities of σS and σD to the RNAP, the concentration of the σS-holoenzyme (henceforth called EσS, with ‘E’ standing for the RNAP core enzyme) would be much smaller than that of EσD even in stationary phase.54 This immediately suggests the need for additional players that modulate σ-factor competition to favour the formation of EσS in the stationary phase. Indeed, several such factors have been identified and described. One such protein, Crl, binds to σS and increases its affinity for the core RNAP, thus favouring the formation of EσS. This promotes the transcription of several σS-dependent genes.55 There is evidence again that the small molecule guanosine tetraphosphate, which signals starvation, also favours alternative σ-factors including σS in its competition with σD for holoenzyme formation.56
Two other molecules influence σ-factor competition by negatively targeting σD and EσD activity. One is the protein Rsd, whose level increases during slow growth,57 including in the stationary phase.58 This protein binds to σD and sequesters it away from the core RNAP, thus removing a proportion of σD from the σ-factor competition. This should favour EσS formation during the stationary phase. The other molecule influencing σ-factor competition is a non-coding RNA called 6S RNA.59 6S RNA is produced by the transcription of a gene called ssrS, but the RNA is not translated into protein. The 6S RNA adopts a looped structure that forms a base-paired motif resembling a standard E. coli promoter. This attracts EσD, thus reducing its availability for transcription while also removing part of σD during the stationary phase when the 6S RNA level is at its highest. Thus, while Rsd removes σD from the equation, 6S RNA reduces the availability of EσD for transcription. The effect of 6S RNA therefore would also reduce the amounts of core RNAP available for σ-factors to bind to. Avantika Lal in my lab asked what effect each of these two methods of modulating the partitioning of transcription space across different Eσ holoenzymes would have on global gene expression in E. coli.60 She used a combination of genome-scale gene expression measurements and mathematical modelling of σ-factor competition to approach this.
Experiments measuring mRNA expression levels of genes in E. coli cells lacking Rsd, in comparison with those that have the protein, showed that very few genes changed substantially in their expression between these two conditions. However, many σS target genes showed small but consistent decreases in expression levels in the absence of Rsd. Though Rsd operates by sequestering σD, σD targets did not show any change in their mRNA levels when Rsd was removed from the system. On the other hand, deletion of 6S RNA resulted in large changes in the expression levels of several genes, but the sets of genes responding to 6S RNA availability depended on the growth phase that the cells were in. But like the Rsd deletion, removal of 6S RNA also caused decreases in the expression of many σS target genes. In contrast to the Rsd deletion, removal of 6S RNA caused an increase in the expression of several σD target genes. Many of these σD targets showing elevated expression in the absence of 6S RNA were expressed at below average levels in the presence of 6S RNA. This suggests that one role for 6S RNA is in suppressing the expression of genes with relatively weak promoters. The removal of both 6S RNA and Rsd produced much greater effects on gene expression than would be expected from the product of these individual deletions.
These findings raised some important questions. First, Rsd acts by sequestering σD and any effect it has on σS function should be indirect. Yet, the aforementioned gene expression experiments showed that the removal of Rsd has an effect on the expression of σS targets but little, if any, on genes regulated by σD. How does this work? To answer this, Lal and colleagues used a mathematical model of holoenzyme formation and transcription that incorporated core RNAP, σD, σS, Rsd, 6S RNA, and DNA (Fig. 5.6). This model showed that in the presence of 6S RNA, increasing Rsd concentration results in the freeing up of EσD from its complex with 6S RNA, leading to the release of some core polymerase which can then bind to σS and form EσS. The end result is an increase in transcription of EσS-dependent promoters. The model also showed that an increase in Rsd concentration also causes a decrease in transcription of σD target genes, something that was not apparent in the gene expression data. The gene expression experiments however showed that Rsd regulates 6S RNA: when Rsd is removed, there is a ~2.5-fold increase in the expression of 6S RNA. This could potentially decrease the availability of EσD for transcription. Incorporation of this regulation of 6S RNA by Rsd into the mathematical model showed that it preserves the effect of Rsd on σS-dependent transcription but abolishes its effect on σD-dependent gene expression.
The second question that arises pertains to the effect of 6S RNA on transcription. 6S RNA not only sequesters σD but, by binding to EσD, also reduces the amount of the core RNAP available for σS to interact with. This should result in an overall decrease in transcription, though this effect would be more pronounced for σD-dependent genes. The theoretical model also supports this view. How then does the removal of 6S RNA selectively decrease σS-dependent transcription while increasing the expression of many σD-dependent genes? The gene expression data showed that the absence of 6S RNA reduced the expression of Rsd and increased that of σS itself. Further, it also decreased the expression of the catalytic subunit of the RNAP, which reduces the overall availability of this enzyme. These effects together help reduce σS-dependent transcription; in fact, a ~20% reduction in Rsd appears to be sufficient to reduce σS-dependent transcription in the absence of 6S RNA. 6S RNA also appears to affect the expression of other regulators of σ-factor competition such as Crl, all of which should contribute to σS competing effectively with σD for binding to core RNAP.
Thus, Rsd and 6S RNA regulate each other and also other players involved in transcription and σ-factor competition. These combined effects appear to be necessary for these molecules to modulate σ-factor competition in a manner that favours σS-dependent gene expression. Why such a complex web of interactions to modulate the interplay between σD and σS? We will try to answer this question when we explore how transcription regulatory networks evolve a little later in this chapter.
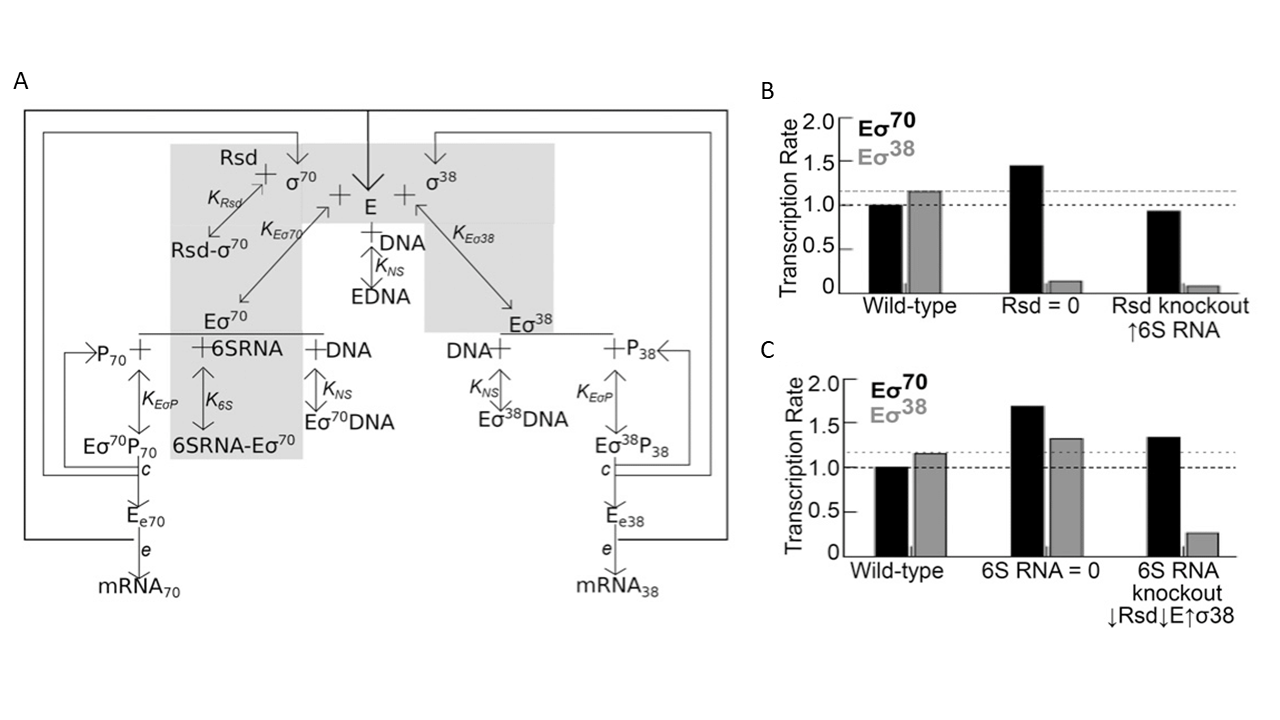
Fig. 5.6. Complex regulation of σS-σD competition. (A) A schematic showing the set of interactions involved in the regulation of σS-σD competition by the protein Rsd and the non-coding RNA 6S RNA. (B) Figure shows calculated transcription rate when Rsd is removed and when 6S RNA levels are increased in the absence of Rsd. (C) Figure shows calculated transcription rate when 6S RNA is removed, and the effect of the observed decrease in Rsd and core RNAP and increase in σS in the absence of 6S RNA. (B) and (C) show how functional connections between 6S RNA and Rsd appear to be important in determining the outcome of the competition between σS and σD. Originally published as part of Figure 6 in A. Lal, S. Krishna, and A.S.N. Seshasayee, ‘Regulation of Global Transcription in Escherichia coli by Rsd and 6S RNA’, Genes Genomes Genetics 8 (2018), 2079–2089, CC BY 4.0.
5.4. Managing the costs of horizontally acquired DNA: the ‘genome sentinel’61
Horizontal transfer is a major mode of gene acquisition in bacteria (see Chapter 4), leading to genome expansion. As with any other segment of DNA, whether a piece of horizontally acquired DNA is maintained in a genome is a function of the selective pressure in its favour. This is especially so in bacteria with such large population sizes that the cost of merely maintaining and expressing a non-functional or neutral piece of DNA is enough for this DNA to be lost from the population (see Chapter 3). As described in Chapter 4, the selective pressure in favour of maintaining a piece of DNA in a genome could be conventional in that the DNA includes genes that enhance the growth and survival of the organism in its niche—for example, by allowing it to resist antibiotics in an antibiotic-rich environment. Or selection could arise from addiction, in which the loss of a piece of DNA once acquired proves toxic to the cell—a concept demonstrated by what are called toxin-antitoxin systems, which are often horizontally acquired. The cost that a piece of DNA presents to its host arises from the metabolic expense of maintaining it, as well as the possibility that its expression could prove toxic to the cell or interfere with the functioning of molecules already well-established in the host. For example, Rotem Sorek and colleagues analysed gene fragments, from nearly 80 prokaryotic genomes, that could not be successfully transferred into E. coli.62 They presented evidence that the expression of such genes, even at low levels, could not be tolerated by the host cell, arguing that these genes are toxic to E. coli. This toxicity prevents their successful establishment in the E. coli genome. For other genes, their failure to transfer into E. coli appeared to be best explained by increased dosage—or, in other words, their high expression levels. That high gene expression is a barrier to horizontal transfer of some genes was reaffirmed very recently by Rama Bhatia and colleagues, who studied the transfer of genes from E. coli into the closely-related Salmonella.63 Thus, both toxicity and inappropriately high expression levels can act as barriers to horizontal gene transfer.
Many organisms encode dedicated mechanisms to control or even ‘silence’ horizontally-acquired genes at the level of their expression. A prominent example among eukaryotes is the silencing of selfish transposable elements by small regulatory RNA molecules in many plants. An example in bacteria involves a TF called H-NS, which in E. coli and related bacteria represses the expression of a variety of horizontally-acquired genes. H-NS, best known for its DNA-binding activities,64 recognises A+T-rich sequences and binds extensively to such stretches of DNA. While doing so, it can form highly rigid or tightly looped structures that can either block the binding of RNAP to promoters or the relative movement of DNA and RNAP when the latter is already bound to a promoter. Now, it turns out that many horizontally-acquired genes in E. coli and related free-living bacteria tend to be A+T-rich. The genomes of E. coli and many related bacteria are usually nearly 50% A+T on average. However, the distribution of the A+T content of genes in these genomes is skewed towards the right. This means that very few genes are G+C-rich, but a larger proportion are A+T-rich, or more A+T-rich than the mean. Many such genes are believed, with good reasons, to have been acquired horizontally. These are often poorly conserved even across closely-related strains and species, and also include genes of bacteriophage origin. The high A+T content of many horizontally-acquired genes makes them attractive to H-NS for binding. Where H-NS binds, it represses transcription. Therefore, H-NS, which preferentially binds A+T-rich genes, emerges as a ‘silencer’ or repressor of the transcription of horizontally-acquired genes.
H-NS, as a protein that modifies the structure of DNA and also regulates gene expression, has been known since the 1980s.65 However, its major role as a silencer of horizontally-acquired genes was not recognised until 2006, when two papers described the binding of H-NS to the chromosome of Salmonella—a close relative of E. coli—and the impact this binding has on gene expression on a genomic scale. The two pieces of work, one by William Navarre et al.66 and the other by Sacha Lucchini et al.,67 showed—by isolating and identifying chromosomal DNA regions bound by H-NS in Salmonella cells—that H-NS binds to hundreds of genes, including many coding for proteins that help the bacteria cause disease (Fig. 5.7). When H-NS was removed from cells by genetic means, genes bound by the protein greatly increased in expression, showing that H-NS represses the expression of genes it binds to. Many genes bound and regulated by H-NS are poorly conserved, are often specific to Salmonella, and show higher A+T-content than is typical of the average gene in this bacterium. Many of these genes have a role to play in the virulence of Salmonella and are normally expressed only during specific stages of infection and not during normal growth. The uncontrolled expression of these virulence-associated genes in the absence of H-NS is detrimental to the host bacterium. In fact, in the absence of additional, compensating mutations in the stress responsive σ-factor σS, the removal of H-NS is lethal to Salmonella. These findings clearly emphasised the importance of gene silencing to the fitness and evolutionary success of these bacteria.
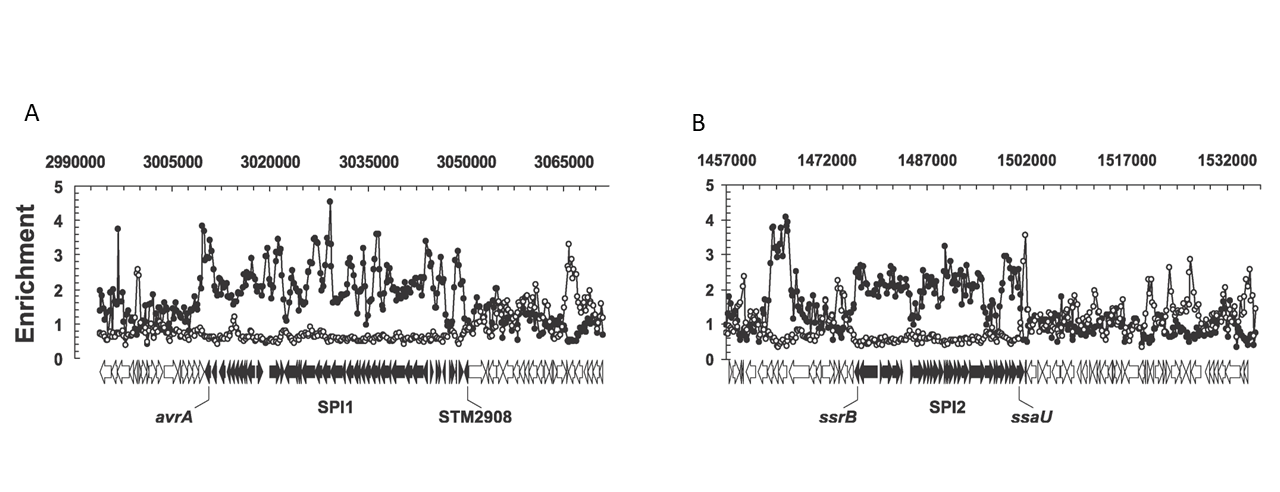
Fig. 5.7. Binding of H-NS to virulence genes. This figure shows the binding of H-NS (enrichment, on the y-axis) to pathogenicity-determining genes belonging to (A) SPI-1 and (B) SPI-2 pathogenicity islands in Salmonella enterica Typhimurium. Originally published as Figures 3D and 3E in S. Lucchini, G. Rowley, M.D. Goldberg, D. Hurd, M. Harrison, and J.C. Hinton, ‘H-NS Mediates the Silencing of Laterally Acquired Genes in Bacteria’, PLoS Pathogens 2 (2006), e81, Creative Commons Attribution License.
A few years later, Christina Kahramanoglou and colleagues, in a piece of work I was involved in, showed—using genome-scale techniques again—that H-NS binds to A+T-rich horizontally-acquired genes in E. coli and represses their expression.68 However, unlike typical TFs, H-NS binding regions on the DNA extend over long stretches. The longer the stretch of DNA bound by H-NS, the more likely that a gene located at or proximal to the bound DNA would be transcriptionally silenced. Thus, the silencing of horizontally-acquired genes by H-NS requires many molecules of the protein to bind adjacently, essentially covering long stretches of DNA like beads on a string.
Studies of H-NS in E. coli, unlike those in Salmonella, rarely reported lethality when the protein was removed. Some studies observed a slight reduction in growth rates, and others none. Despite performing similar functions in Salmonella and E. coli, H-NS is essential for bacterial survival in one and not the other. These observations suggest that there is something else in the genome of E. coli, presumably differing in some way from the contents of the Salmonella genome, that minimises the impact of the loss of H-NS on survival. What might this be? The E. coli genome encodes a protein called StpA, which is similar in sequence to H-NS. StpA also binds to A+T-rich DNA sequences, but is produced by E. coli cells in much smaller quantities than H-NS. Removing StpA from E. coli cells has little, if any, impact on growth, at least in laboratory conditions. Ebru Uyar and colleagues identified sites on the chromosome within E. coli cells that are bound by H-NS and StpA in the presence and absence of the other protein.69 They first found that the binding of H-NS to the E. coli chromosome is unaffected by the presence or absence of StpA. StpA binds to the same locations as H-NS when the latter is also present. However, when H-NS is removed from cells, StpA loses its ability to bind to as many as two-thirds of its sites. Uyar and co-workers further suggest that this reduction in binding of StpA to the chromosome in the absence of H-NS probably reflects the intrinsic binding properties of StpA. They also propose that H-NS can induce changes in the structure of the DNA that it binds to, which might enable StpA to bind to these regions of the chromosome. The loss of such DNA structural features in the absence of H-NS might reduce the ability of StpA to bind to it.
Does the binding of StpA to a small subset of its targets in the absence of H-NS help maintain the repression of these genes? If so, is there some basis to which subset of H-NS regulated genes are ‘chosen’ for repression by the StpA-dependent backup regulatory system? Rajalakshmi Srinivasan in my lab attempted to address this question. She first asked what effect the removal of one or both proteins will have on the expression levels of horizontally-acquired genes in E. coli.70 Consistent with the findings of Uyar and colleagues, she found that loss of StpA affects the expression of very few genes. However, when H-NS is removed, the expression of many of its targets—including horizontally-acquired genes—greatly increases. Most importantly, when both H-NS and StpA are removed, many other genes—including those bound by H-NS but unaffected by the loss of H-NS alone—increase in expression. Srinivasan and colleagues compared their gene expression data with Uyar and colleagues’ chromosome binding data for H-NS and StpA. In doing so, they found that genes whose expression increases when StpA is removed from E. coli already lacking H-NS are often those to which StpA remains bound in the absence of H-NS.
In an earlier study, Blair Gordon and colleagues had measured the affinity of H-NS to tens of thousands of 8-mer DNA sequences.71 Using these data, Srinivasan and colleagues noted that regions to which StpA binds in the absence of H-NS tend to have a high density of 8-mers that display high-affinity binding to H-NS—and, by inference, to StpA. This finding offers a biophysical rationale for how StpA is able to retain its ability to bind to some but not all its wildtype sites. Srinivasan and co-workers also found that while the loss of H-NS alone has very little effect on growth of E. coli under the conditions they had tested in the lab, the loss of both H-NS and StpA resulted in a large growth impairment. Thus, StpA binds to a subset of H-NS-repressed, horizontally-acquired genes in the absence of H-NS and dampens their over-expression when H-NS is lost from the system. This backup function of StpA also helps soften the adverse effect of the loss of H-NS on bacterial growth. Thus, these results lead to the suggestion that keeping horizontally-acquired genes—to which StpA binds in the absence of H-NS—transcriptionally silent is important to ensure that the loss of H-NS on its own does not severely impair the growth of E. coli.
Srinivasan and co-workers also observed that genes that are repressed by StpA in the absence of H-NS: (a) are expressed at very low levels, lower than genes repressed by H-NS but not by StpA in the absence of H-NS; (b) transition to very high expression levels when H-NS and StpA are removed from the system. These two findings suggest that these horizontally-acquired genes that are silenced by both H-NS and StpA have a high intrinsic ability for transcription. Transcription at these genes may not even produce full length mRNA.72 Instead, the high A+T content of these genes, in the absence of H-NS/StpA, exposes many promoter-like elements within gene sequences. This attracts RNAP, causing it to waste resources by performing useless transcription. Going by the bioenergetic cost calculations by Lynch and Marinov that we discussed in Chapter 3, these genes—if left unregulated—would carry a very large negative selection coefficient (s << 0) and be detrimental to the cell if not eliminated. And the role of the silencing system orchestrated by H-NS and StpA is to ensure that they do not get expressed inappropriately when not required. In fact, as demonstrated by Marie Doyle and co-workers, some horizontally-acquired plasmids—accessory genetic elements found in many copies in cells—encode their own version of H-NS to ensure that the expression of plasmid-borne genes is kept under control without syphoning off the host cell’s endogenous H-NS reserves.73 This function, by ensuring that the use of H-NS to silence plasmid-encoded genes does not compromise its function on chromosomal DNA, enables the maintenance of the plasmid in its host.
Though many horizontally-acquired genes may be detrimental to the cell if allowed to be transcribed under favourable growth conditions, there is evidence that some of these genes might in fact be beneficial (s >> 0) under stress. For example, several genes normally repressed by H-NS in Salmonella are required for virulence, but can negatively impact growth when expressed under benevolent laboratory conditions. Positive s under certain conditions could ensure that these genes are maintained in the bacterial population. Additional regulatory mechanisms, such as anti-H-NS proteins that displace H-NS from the DNA, can relieve repression by H-NS and StpA precisely when necessary.74
Salmonella, in which the deletion of H-NS is lethal, also encodes an StpA. However, it does not seem to be capable of supporting bacterial survival and growth in the absence of H-NS. Why would this be so? Sabrina Ali and colleagues allowed Salmonella lacking H-NS75 to grow in the laboratory in such a way that these slow-growing populations could accumulate additional mutations.76 Some of these mutations would be adaptive, allowing the bearer to grow faster than its parent. Such adaptive mutations would soon dominate in the population, as predicted by natural selection. This would then allow investigators to discover mechanisms by which Salmonella can compensate for growth defects caused by the loss of H-NS. Ali et al. found that the loss of pathogenicity islands—which are usually kept silent by H-NS but are expressed at high levels in an inappropriate manner in the absence of the repressor—allows Salmonella lacking H-NS to adapt to the loss of the repressor. This finding reinforces the idea that improper expression of horizontally-acquired virulence genes, under conditions in which virulence has no role to play in the organism’s lifestyle, can be costly. It also shows that evolution would quickly result in the loss of such expensive pieces of DNA if they happened to reside and be expressed inside bacterial cells when not required. In addition, these researchers found that mutations in StpA also allowed the H-NS-negative Salmonella to adapt to the absence of H-NS (Fig. 5.8A). StpA in Salmonella is not identical in sequence to that in E. coli. A quick look at the two StpA sequences showed that some of the mutations which allowed Salmonella to adapt to the loss of H-NS targeted amino acid positions at which the Salmonella StpA differed from the E. coli StpA.77 An open question from this analysis is whether the Salmonella StpA, in its normal form, is unable to act as an effective backup for H-NS, and whether the mutations discovered by Ali and co-workers allow it to do so!
Rajalakshmi Srinivasan in my lab performed an experiment similar to that by Sabrina Ali and colleagues, but for E. coli lacking both H-NS and StpA.78 She expected to see losses of segments of horizontally-acquired DNA in populations displaying adaptation to the absence of H-NS and StpA. However, this did not happen. Instead, she first observed that mutations that inactivate σS emerged; this was not surprising in light of prior evidence linking H-NS and σS. Yet, this did highlight an important point which we will examine shortly: that mutations which perturb portions of the transcriptional regulatory network can be adaptive.
Fig. 5.8. Compensation for the loss of the H-NS gene silencing system. (A) This figure shows that mutations that change StpA and those that delete clusters of pathogenicity-related genes compensate for the loss of H-NS in Salmonella. Originally published as Figure 3 in S.S. Ali, J. Soo, C. Rao, A.S. Leung, D.H. Ngai, A.W. Ensminger, and W.W. Navarre, ‘Silencing by H-NS potentiated the evolution of Salmonella’, PLoS Pathogens 10 (2014), e1004500, Creative Commons Attribution License. (B) This figure shows that a duplication of nearly 40% of the chromosome centred around ori (which is located at ~3.9e06 on the x-axis) partially compensated for the lack of H-NS and StpA in E. coli. Originally published as Figure 3E in R. Srinivasan, V.F. Scolari, M.C. Lagomarsino, and A.S.N. Seshasayee, ‘The genome-scale interplay amongst xenogene silencing, stress response and chromosome architecture in Escherichia coli’, Nucleic Acids Research 43 (2005), 295–308, CC BY 4.0.
A second mutation that emerged, partly compensating for the loss of H-NS and StpA, was a duplication of nearly 40% of the chromosome (Fig. 5.8B). This mutation, while increasing the expression of many genes in the duplicated segment of the chromosome, also caused a strong reduction in the expression of horizontally-acquired genes that had been derepressed by the loss of H-NS and StpA. This underlined the idea that rearrangements of large parts of the chromosome can also be adaptive, and can compensate in part for the loss of a global regulatory network. Keeping in mind the fact that the H-NS-StpA system primarily represses horizontally-acquired genes, we can now ask the following questions: are genes of different functions and of different evolutionary origins positioned differently on the chromosome, and how does this arrangement interplay with gene expression? We will address these questions in the final section of this book. But before that, we make a detour and ask how transcriptional regulatory networks evolve.